

Common genetic variations in telomere length genes and lung cancer: a Mendelian randomisation study and its novel application in lung tumour transcriptome
- PMID: 37079368
- PMCID: PMC10118386
- DOI: 10.7554/eLife.83118
Common genetic variations in telomere length genes and lung cancer: a Mendelian randomisation study and its novel application in lung tumour transcriptome
Abstract
Background: Genome-wide association studies (GWASs) have identified genetic susceptibility variants for both leukocyte telomere length (LTL) and lung cancer susceptibility. Our study aims to explore the shared genetic basis between these traits and investigate their impact on somatic environment of lung tumours.
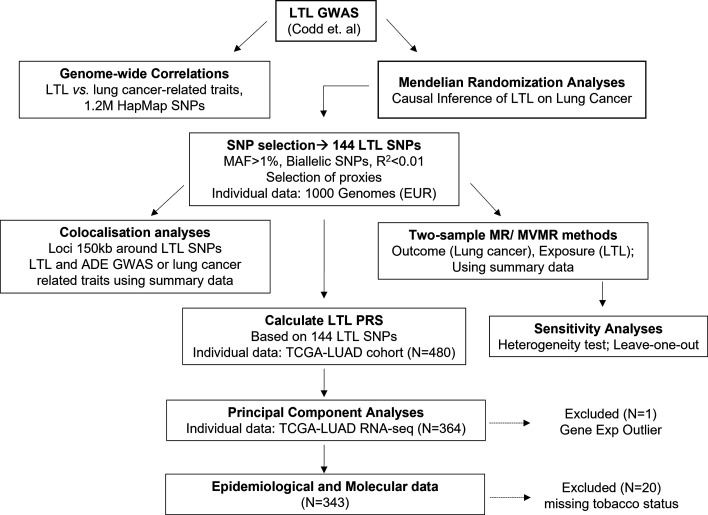
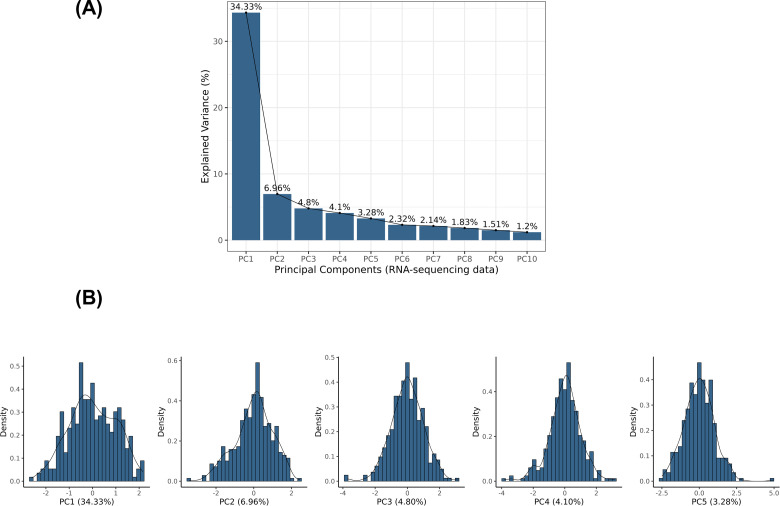
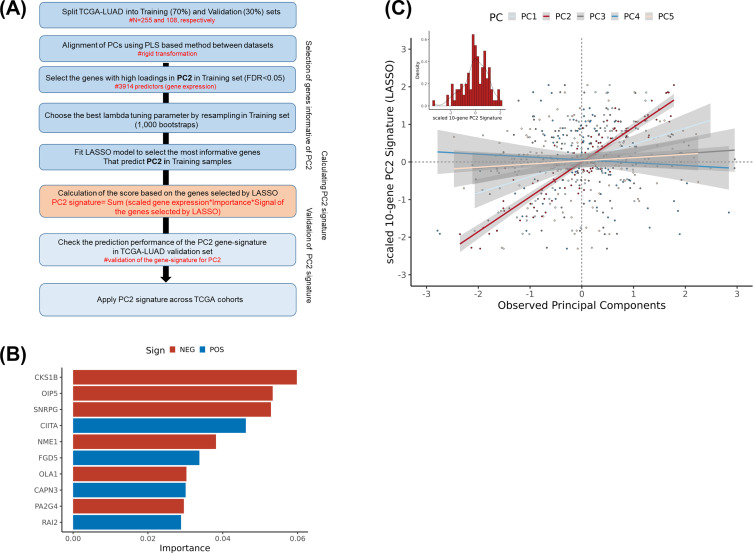
Methods: We performed genetic correlation, Mendelian randomisation (MR), and colocalisation analyses using the largest available GWASs summary statistics of LTL (N=464,716) and lung cancer (N=29,239 cases and 56,450 controls). Principal components analysis based on RNA-sequencing data was used to summarise gene expression profile in lung adenocarcinoma cases from TCGA (N=343).
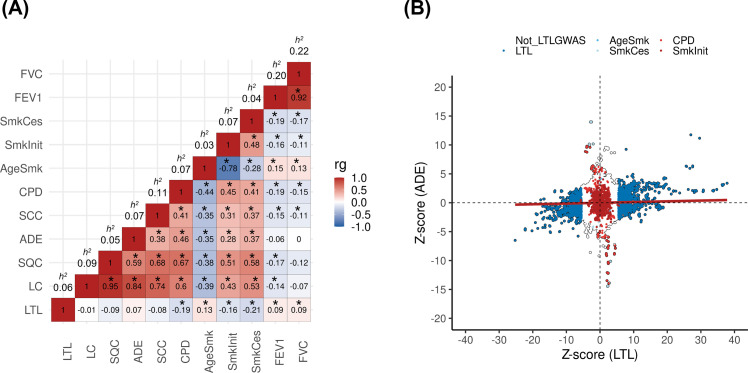
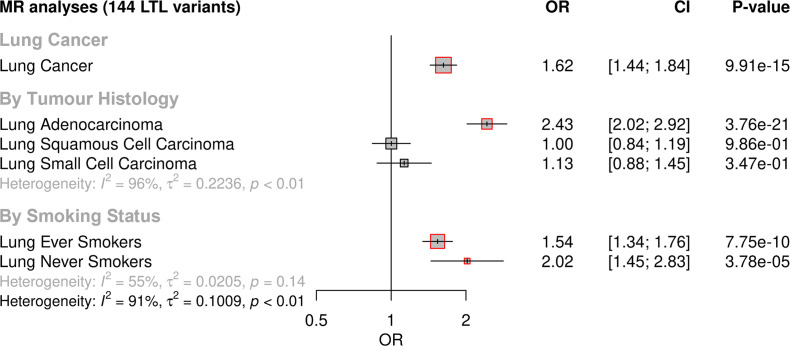
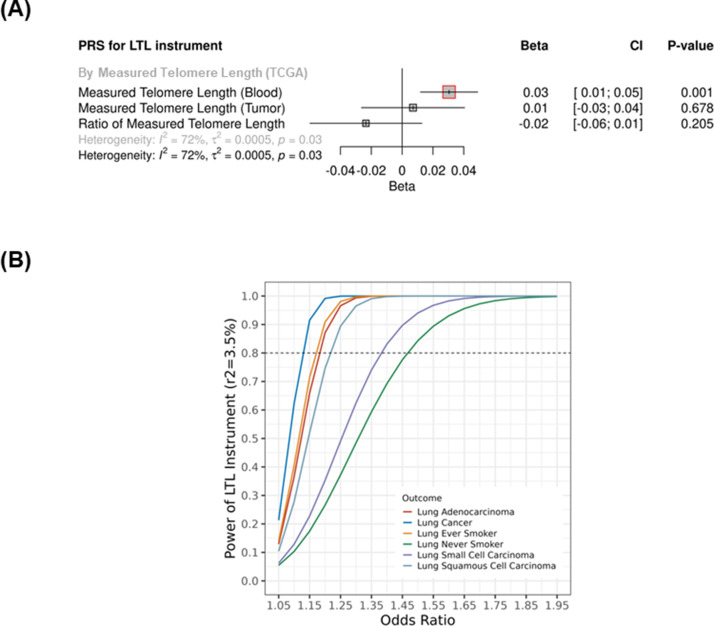
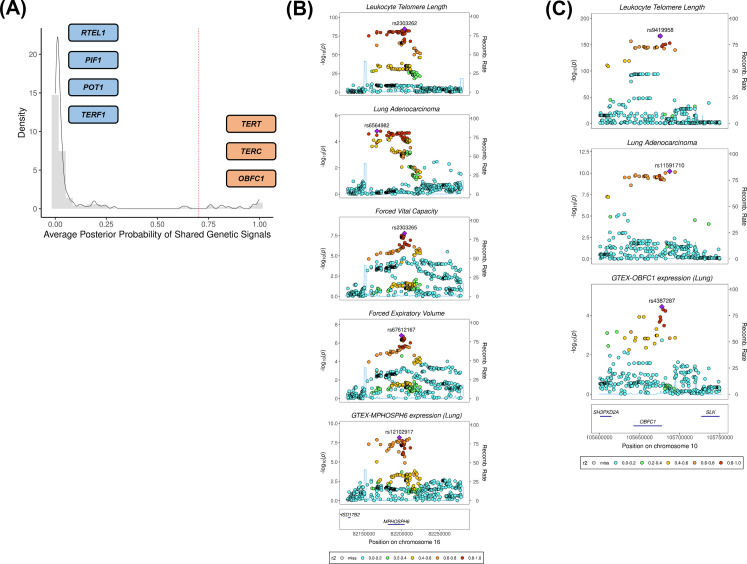
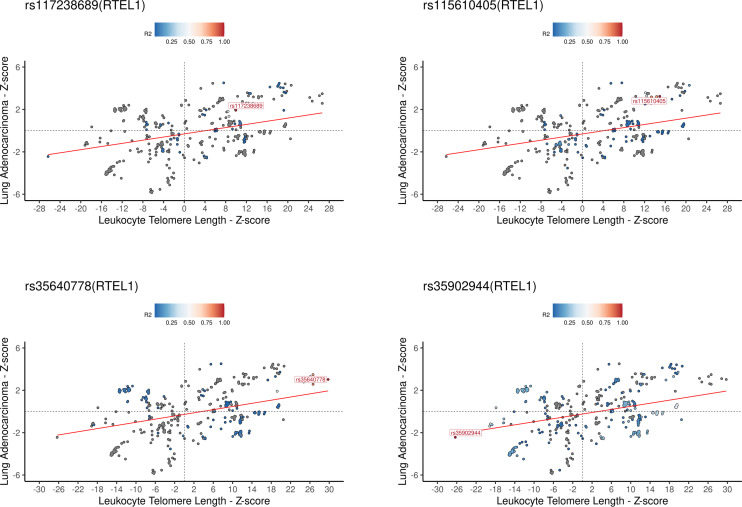
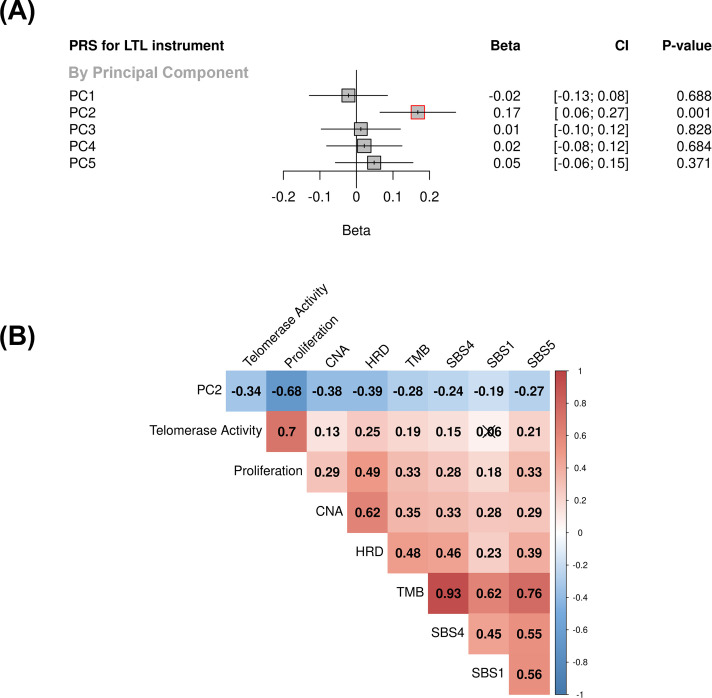
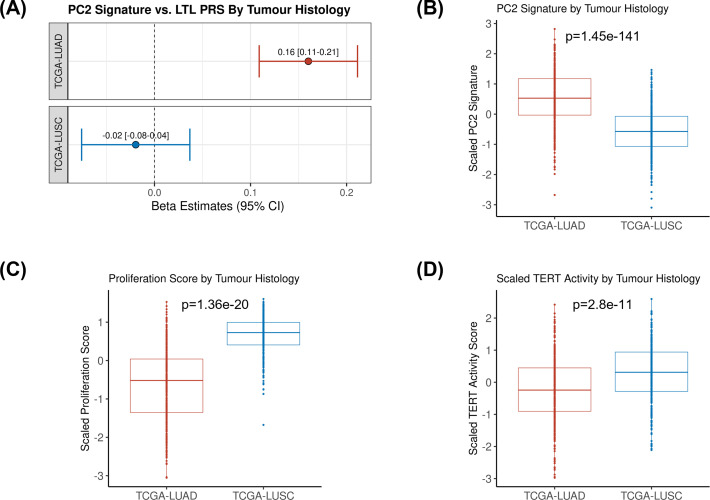
Results: Although there was no genome-wide genetic correlation between LTL and lung cancer risk, longer LTL conferred an increased risk of lung cancer regardless of smoking status in the MR analyses, particularly for lung adenocarcinoma. Of the 144 LTL genetic instruments, 12 colocalised with lung adenocarcinoma risk and revealed novel susceptibility loci, including MPHOSPH6, PRPF6, and POLI. The polygenic risk score for LTL was associated with a specific gene expression profile (PC2) in lung adenocarcinoma tumours. The aspect of PC2 associated with longer LTL was also associated with being female, never smokers, and earlier tumour stages. PC2 was strongly associated with cell proliferation score and genomic features related to genome stability, including copy number changes and telomerase activity.
Conclusions: This study identified an association between longer genetically predicted LTL and lung cancer and sheds light on the potential molecular mechanisms related to LTL in lung adenocarcinomas.
Funding: Institut National du Cancer (GeniLuc2017-1-TABAC-03-CIRC-1-TABAC17-022), INTEGRAL/NIH (5U19CA203654-03), CRUK (C18281/A29019), and Agence Nationale pour la Recherche (ANR-10-INBS-09).
Keywords: GWAS; Genome Stability; Mendelian randomisation; epidemiology; gene expression; global health; human; lung cancer; telomere length.
© 2023, Cortez Cardoso Penha et al.
Conflict of interest statement
RC, KS, JA, PH, SK, VC, NS, CN, MM, AG, CA, PB, RH, LK, JM No competing interests declared
Figures
Update of
- doi: 10.1101/2022.08.24.22279131
Similar articles
-
Shared genetic architecture between leukocyte telomere length and Alzheimer's disease.Alzheimers Res Ther. 2025 May 17;17(1):108. doi: 10.1186/s13195-025-01757-z. Alzheimers Res Ther. 2025. PMID: 40382655 Free PMC article.
-
Associations between Longer Leukocyte Telomere Length and Increased Lung Cancer Risk among Never Smokers in Urban China.Cancer Epidemiol Biomarkers Prev. 2023 Dec 1;32(12):1734-1737. doi: 10.1158/1055-9965.EPI-23-0881. Cancer Epidemiol Biomarkers Prev. 2023. PMID: 37721487 Free PMC article.
-
Predicted leukocyte telomere length and risk of myeloid neoplasms.Hum Mol Genet. 2023 Oct 4;32(20):2996-3005. doi: 10.1093/hmg/ddad126. Hum Mol Genet. 2023. PMID: 37531260 Free PMC article.
-
Long or short? Telomere length and pancreatic cancer and its precursor lesions, a narrative review.Mutagenesis. 2025 Mar 15;40(1):39-47. doi: 10.1093/mutage/gead034. Mutagenesis. 2025. PMID: 37976300 Review.
-
Association between telomere length and mitochondrial copy number and cancer risk in humans: A meta-analysis on more than 300,000 individuals.Crit Rev Oncol Hematol. 2021 Nov;167:103510. doi: 10.1016/j.critrevonc.2021.103510. Epub 2021 Oct 22. Crit Rev Oncol Hematol. 2021. PMID: 34695574 Review.
Cited by
-
Multi-omics identify ribosome related causal genes methylation, splicing, and expression in prostate cancer.Discov Oncol. 2025 May 12;16(1):740. doi: 10.1007/s12672-025-02584-2. Discov Oncol. 2025. PMID: 40354008 Free PMC article.
-
Relationship Between Mitochondrial Biological Function and Breast Cancer: An Approach Based on Mendelian Randomization Analysis.Breast J. 2024 Nov 6;2024:4434466. doi: 10.1155/2024/4434466. eCollection 2024. Breast J. 2024. PMID: 39742384 Free PMC article.
-
Evidence based on Mendelian randomization: Causal relationship between mitochondrial biological function and lung cancer and its subtypes.Neoplasia. 2023 Dec;46:100950. doi: 10.1016/j.neo.2023.100950. Epub 2023 Nov 16. Neoplasia. 2023. PMID: 37976568 Free PMC article.
-
Leukocyte telomere length decreased the risk of mortality in patients with alcohol-associated liver disease.Front Endocrinol (Lausanne). 2024 Dec 12;15:1462591. doi: 10.3389/fendo.2024.1462591. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 39735642 Free PMC article.
-
Genomic instability in congenital lung malformations in children.Pediatr Surg Int. 2024 Sep 5;40(1):248. doi: 10.1007/s00383-024-05835-5. Pediatr Surg Int. 2024. PMID: 39237666
References
-
- Adler AS, McCleland ML, Yee S, Yaylaoglu M, Hussain S, Cosino E, Quinones G, Modrusan Z, Seshagiri S, Torres E, Chopra VS, Haley B, Zhang Z, Blackwood EM, Singh M, Junttila M, Stephan JP, Liu J, Pau G, Fearon ER, Jiang Z, Firestein R. An integrative analysis of colon cancer identifies an essential function for prpf6 in tumor growth. Genes & Development. 2014;28:1068–1084. doi: 10.1101/gad.237206.113. - DOI - PMC - PubMed
-
- Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, Akdemir KC, Seth S, Song X, Wang Q, Lichtenberg T, Hu J, Zhang J, Zheng S, Verhaak RGW. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nature Genetics. 2017;49:349–357. doi: 10.1038/ng.3781. - DOI - PMC - PubMed
-
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P, Marchini J. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
