

HSV-2 triggers upregulation of MALAT1 in CD4+ T cells and promotes HIV latency reversal
- PMID: 37079384
- PMCID: PMC10232005
- DOI: 10.1172/JCI164317
HSV-2 triggers upregulation of MALAT1 in CD4+ T cells and promotes HIV latency reversal
Abstract
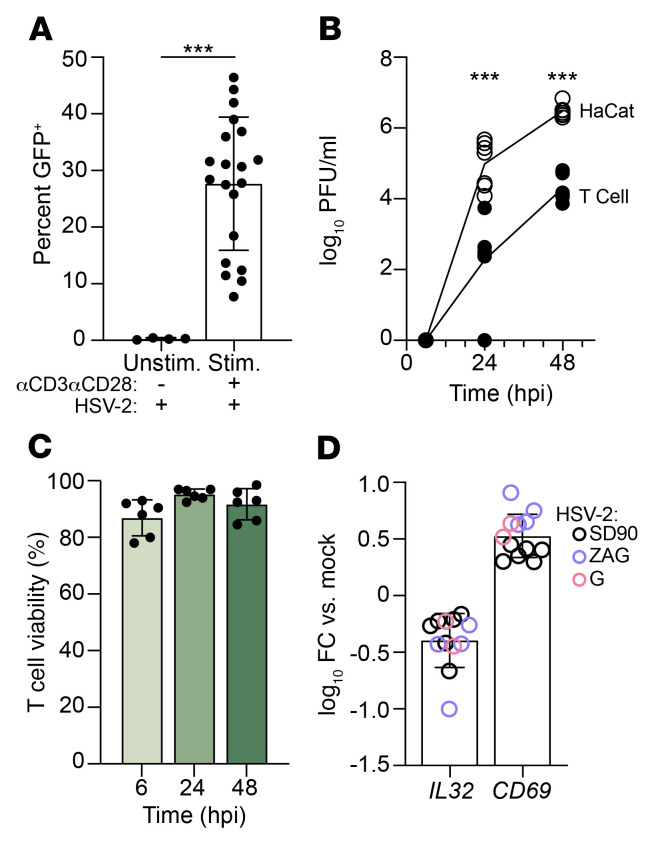
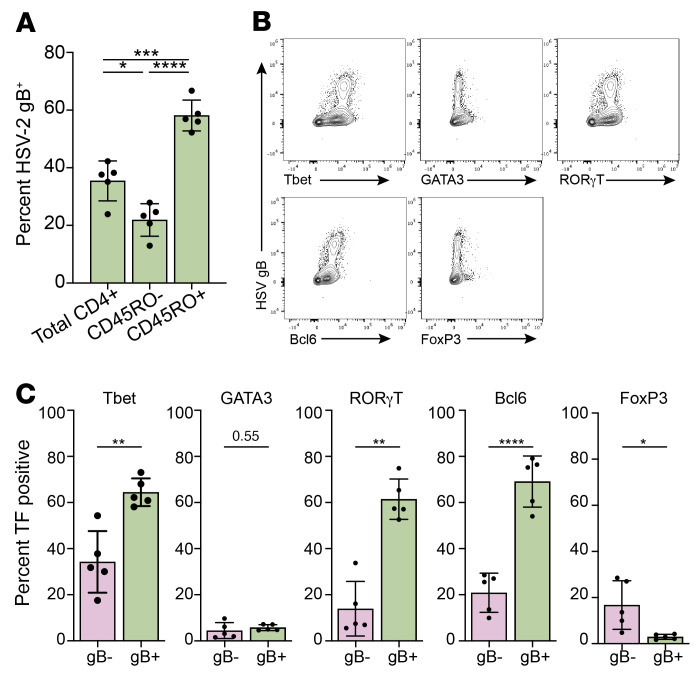
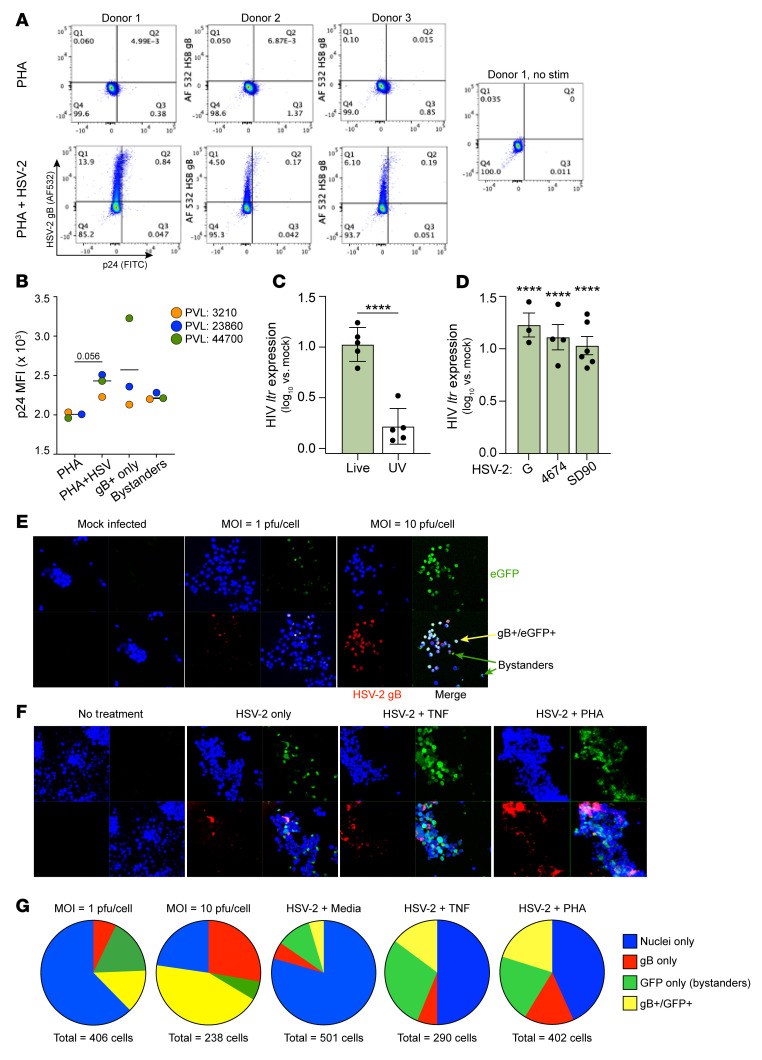
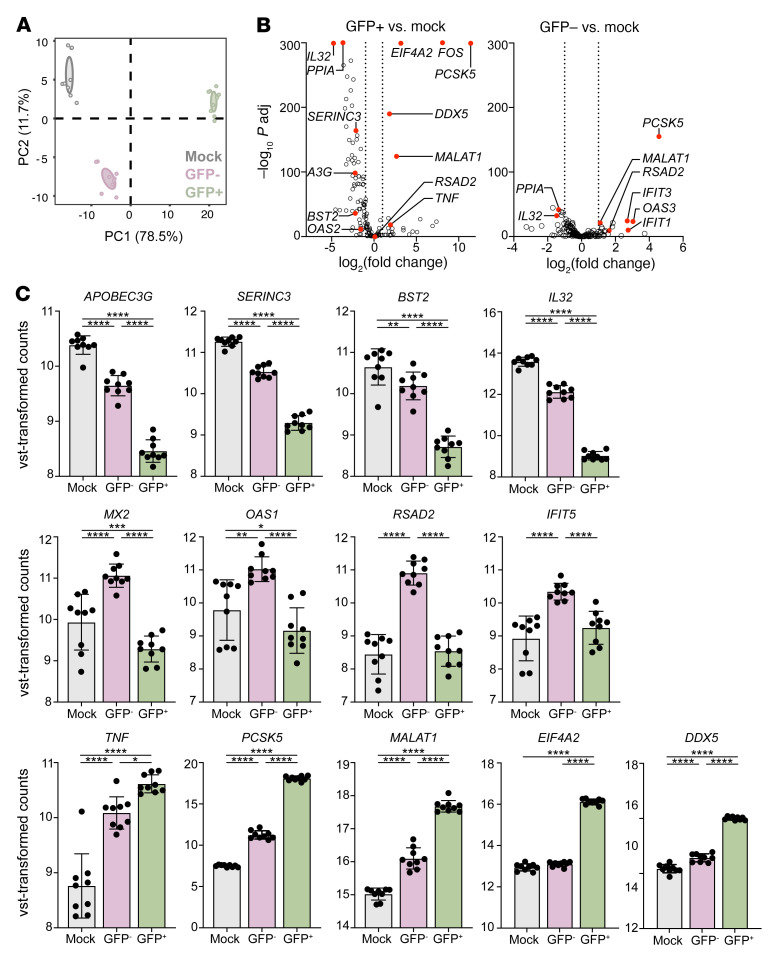
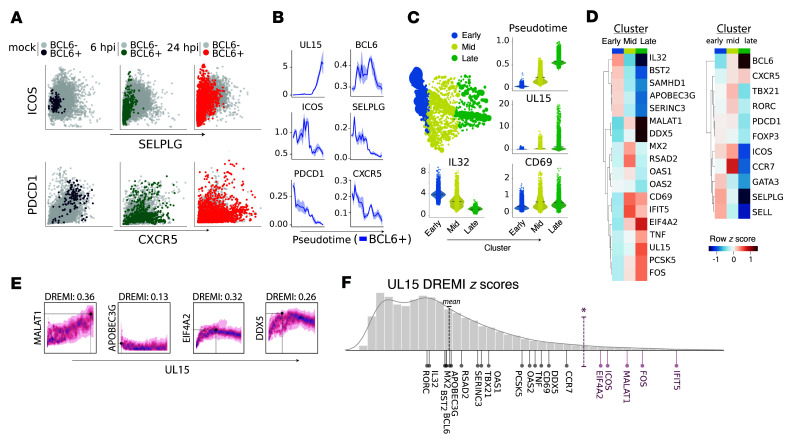
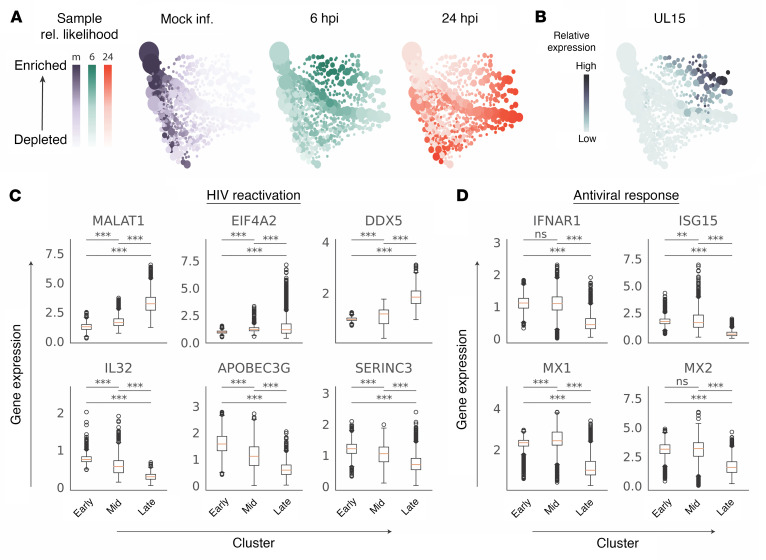
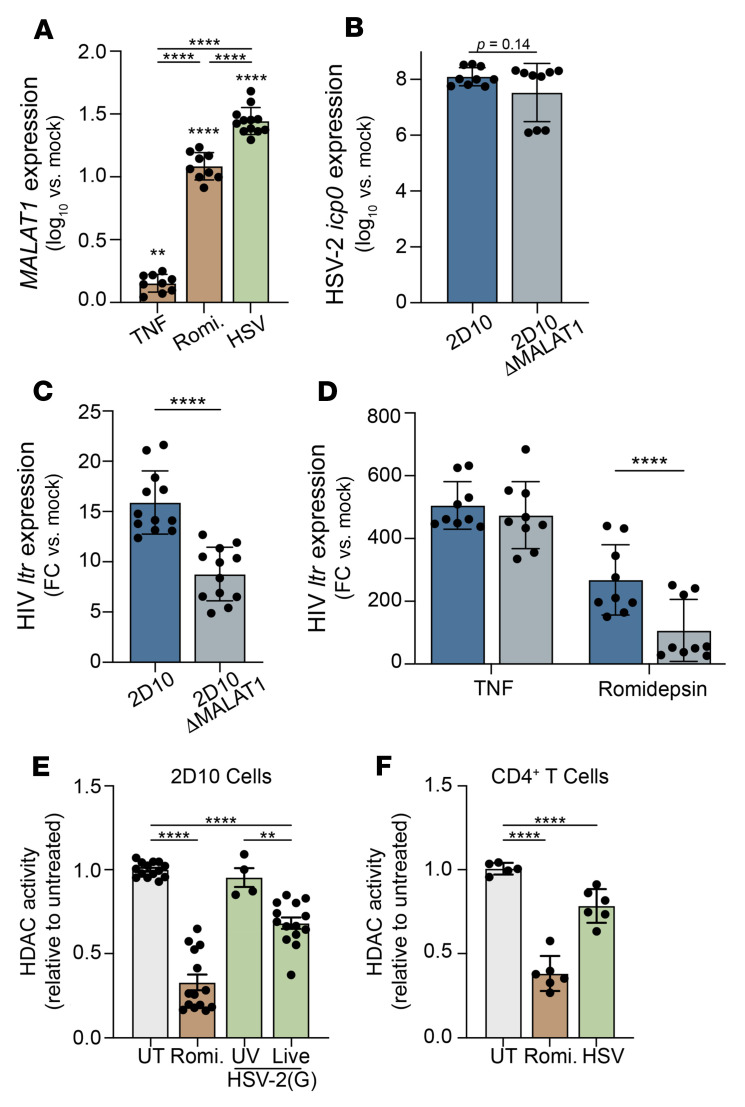
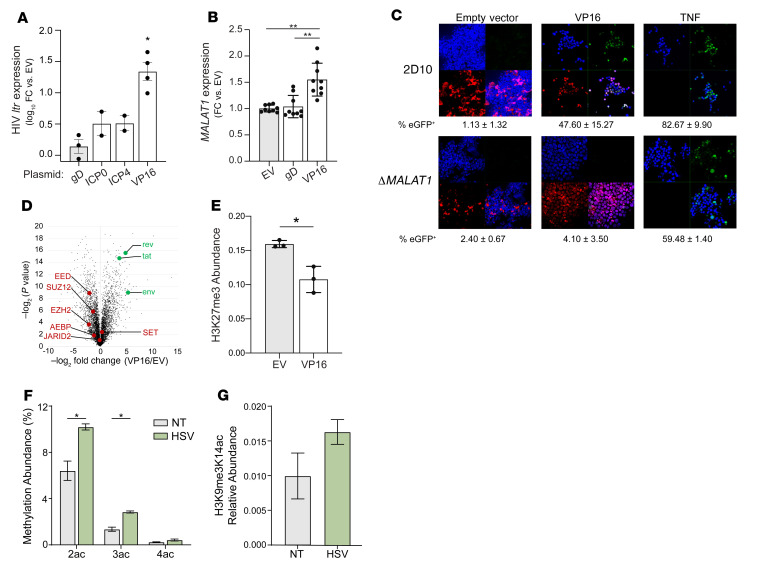
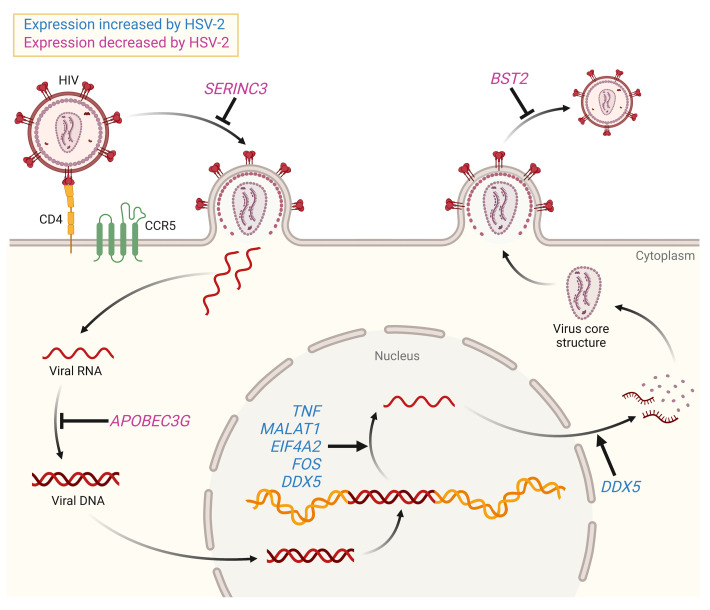
Herpes simplex virus type 2 (HSV-2) coinfection is associated with increased HIV-1 viral loads and expanded tissue reservoirs, but the mechanisms are not well defined. HSV-2 recurrences result in an influx of activated CD4+ T cells to sites of viral replication and an increase in activated CD4+ T cells in peripheral blood. We hypothesized that HSV-2 induces changes in these cells that facilitate HIV-1 reactivation and replication and tested this hypothesis in human CD4+ T cells and 2D10 cells, a model of HIV-1 latency. HSV-2 promoted latency reversal in HSV-2-infected and bystander 2D10 cells. Bulk and single-cell RNA-Seq studies of activated primary human CD4+ T cells identified decreased expression of HIV-1 restriction factors and increased expression of transcripts including MALAT1 that could drive HIV replication in both the HSV-2-infected and bystander cells. Transfection of 2D10 cells with VP16, an HSV-2 protein that regulates transcription, significantly upregulated MALAT1 expression, decreased trimethylation of lysine 27 on histone H3 protein, and triggered HIV latency reversal. Knockout of MALAT1 from 2D10 cells abrogated the response to VP16 and reduced the response to HSV-2 infection. These results demonstrate that HSV-2 contributes to HIV-1 reactivation through diverse mechanisms, including upregulation of MALAT1 to release epigenetic silencing.
Keywords: AIDS/HIV; T cells; Transcription; Virology.
Figures
Similar articles
-
Long noncoding RNA MALAT1 releases epigenetic silencing of HIV-1 replication by displacing the polycomb repressive complex 2 from binding to the LTR promoter.Nucleic Acids Res. 2019 Apr 8;47(6):3013-3027. doi: 10.1093/nar/gkz117. Nucleic Acids Res. 2019. PMID: 30788509 Free PMC article.
-
Subclinical Genital Herpes Shedding in HIV/Herpes Simplex Virus 2-Coinfected Women during Antiretroviral Therapy Is Associated with an Increase in HIV Tissue Reservoirs and Potentially Promotes HIV Evolution.J Virol. 2020 Dec 9;95(1):e01606-20. doi: 10.1128/JVI.01606-20. Print 2020 Dec 9. J Virol. 2020. PMID: 33028713 Free PMC article.
-
MALAT1 is important for facilitating HIV-1 latency reversal in latently infected monocytes.Gene. 2025 Feb 5;936:149095. doi: 10.1016/j.gene.2024.149095. Epub 2024 Nov 15. Gene. 2025. PMID: 39549778
-
A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation.J Gen Virol. 2015 Jul;96(Pt 7):1581-602. doi: 10.1099/vir.0.000128. Epub 2015 Mar 20. J Gen Virol. 2015. PMID: 25794504 Free PMC article. Review.
-
Immunity in latent Herpes simplex virus infection.Acta Virol. 2005;49(3):159-67. Acta Virol. 2005. PMID: 16178513 Review.
Cited by
-
Prospects for therapeutic T-cell vaccine strategies for HIV cure.Curr Opin HIV AIDS. 2025 Sep 1;20(5):463-471. doi: 10.1097/COH.0000000000000965. Epub 2025 Jul 9. Curr Opin HIV AIDS. 2025. PMID: 40638102 Free PMC article. Review.
-
Immunogenicity and Protective Efficacy of an mRNA Vaccine Targeting HSV-2 UL41 in Mice.Vaccines (Basel). 2025 Mar 5;13(3):271. doi: 10.3390/vaccines13030271. Vaccines (Basel). 2025. PMID: 40266094 Free PMC article.
-
A targeted CRISPR screen identifies ETS1 as a regulator of HIV-1 latency.PLoS Pathog. 2025 Apr 8;21(4):e1012467. doi: 10.1371/journal.ppat.1012467. eCollection 2025 Apr. PLoS Pathog. 2025. PMID: 40198713 Free PMC article.
-
Evaluating anemia in HIV-infected patients using chest CT.Open Med (Wars). 2024 Jul 11;19(1):20240996. doi: 10.1515/med-2024-0996. eCollection 2024. Open Med (Wars). 2024. PMID: 39006953 Free PMC article.
-
Purging viral latency by a bifunctional HSV-vectored therapeutic vaccine in chronically SIV-infected macaques.Elife. 2025 Apr 23;13:RP95964. doi: 10.7554/eLife.95964. Elife. 2025. PMID: 40266253 Free PMC article.
