

Induction of lysosomal and mitochondrial biogenesis by AMPK phosphorylation of FNIP1
- PMID: 37079666
- PMCID: PMC10794112
- DOI: 10.1126/science.abj5559
Induction of lysosomal and mitochondrial biogenesis by AMPK phosphorylation of FNIP1
Abstract
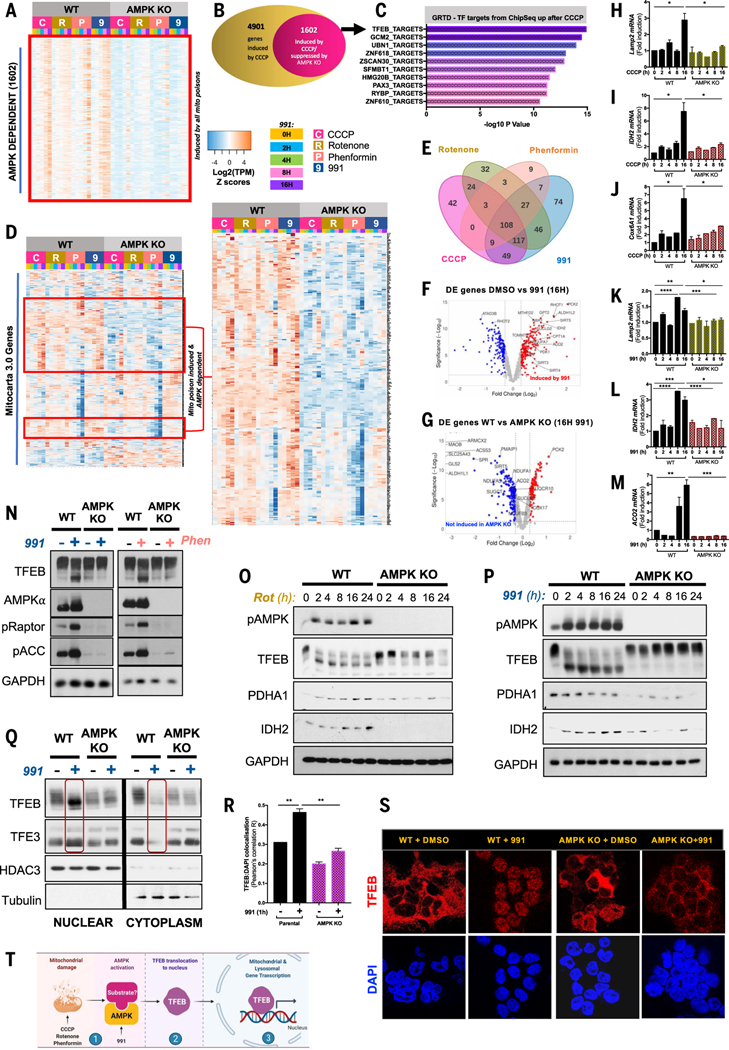
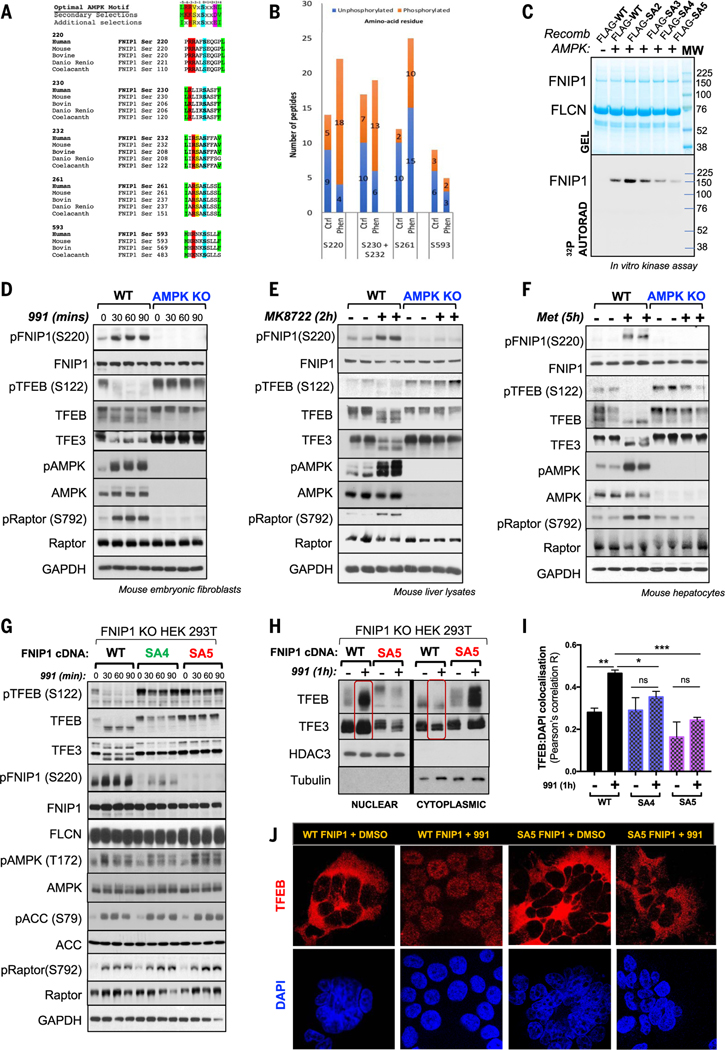
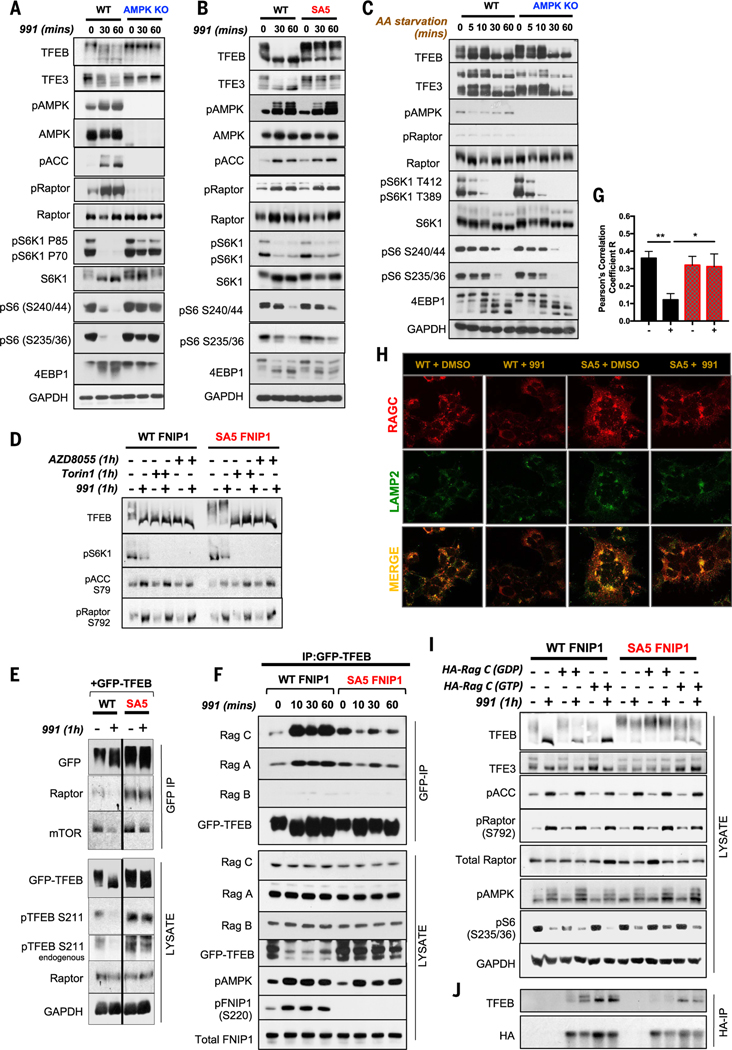
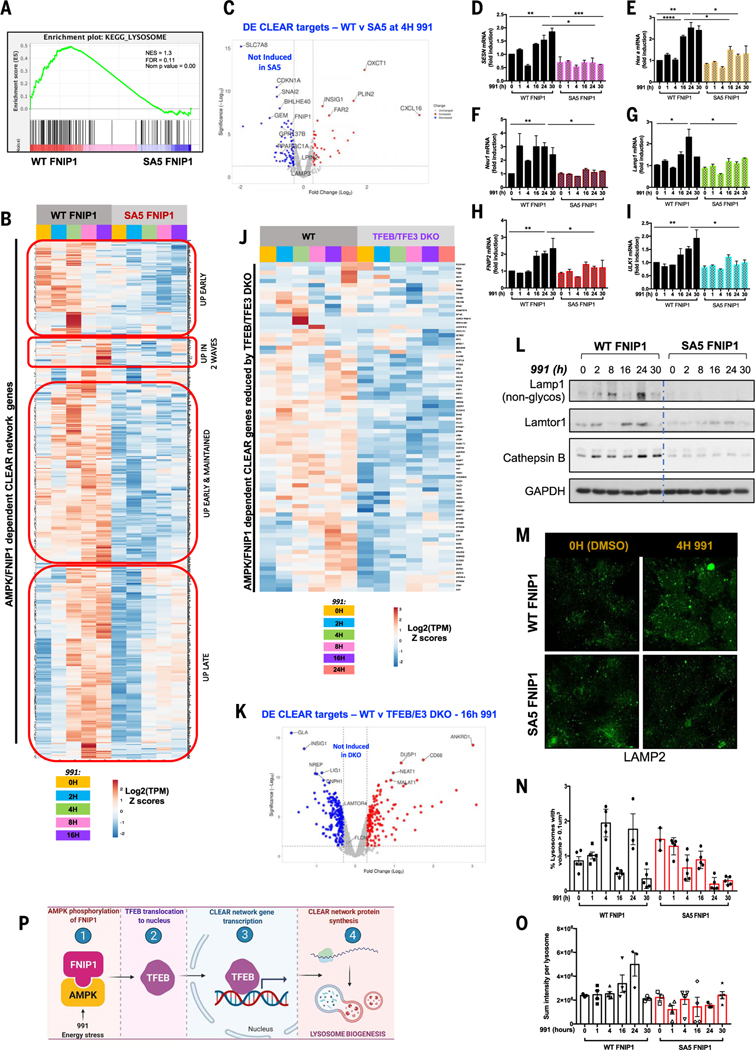
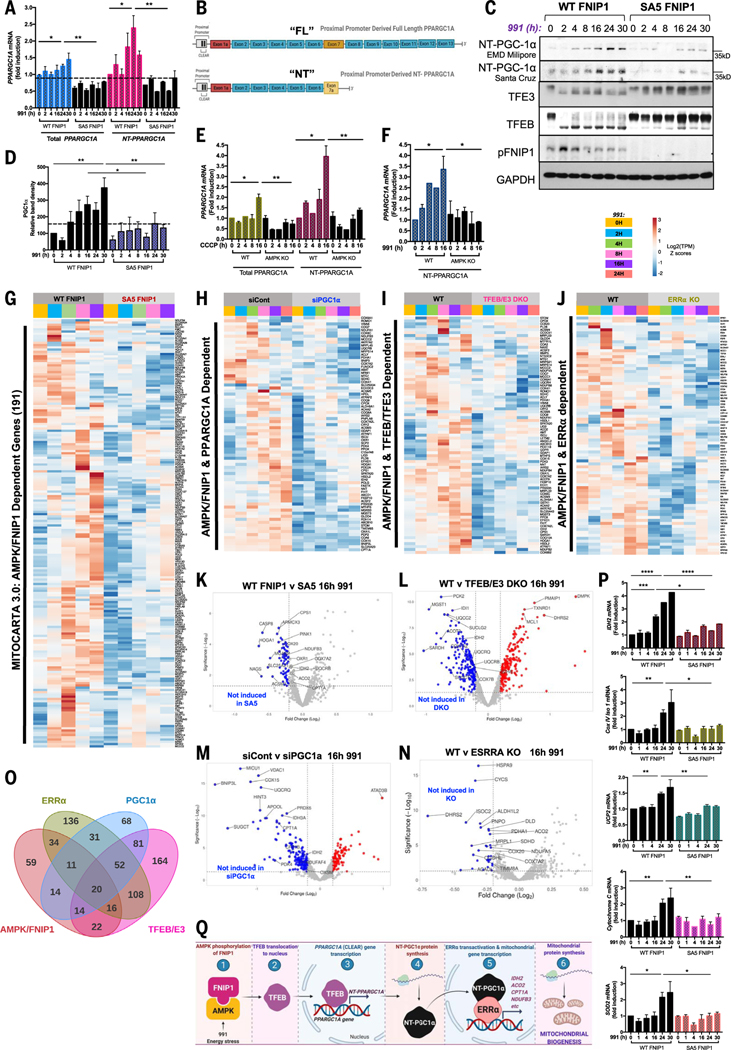
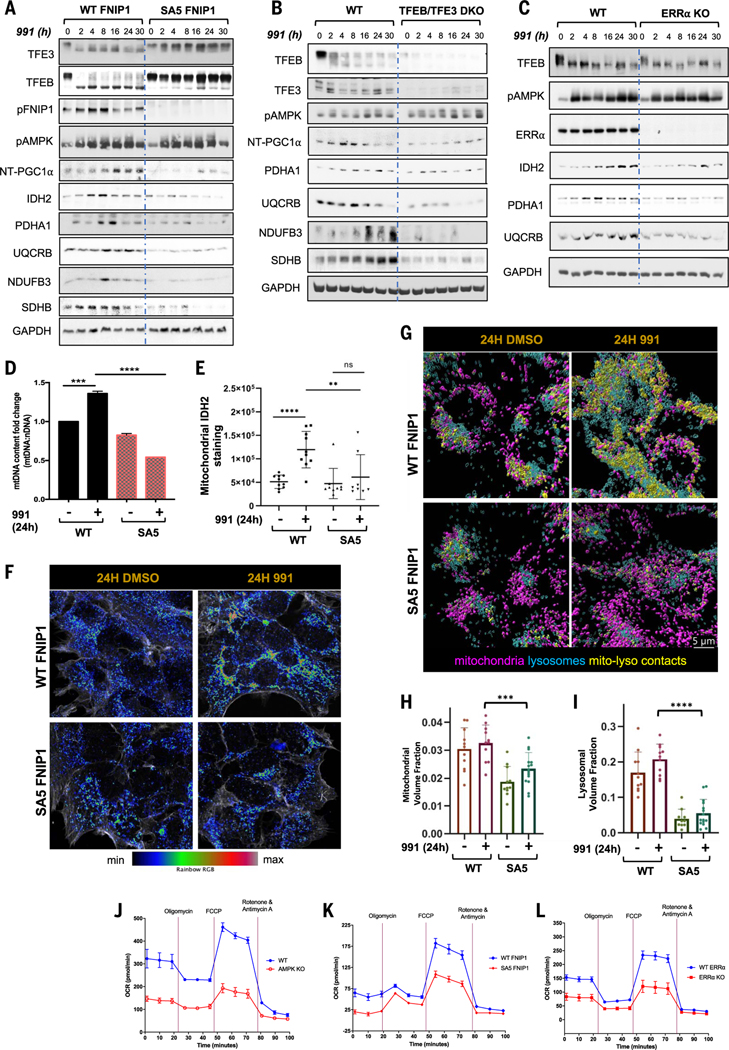
Cells respond to mitochondrial poisons with rapid activation of the adenosine monophosphate-activated protein kinase (AMPK), causing acute metabolic changes through phosphorylation and prolonged adaptation of metabolism through transcriptional effects. Transcription factor EB (TFEB) is a major effector of AMPK that increases expression of lysosome genes in response to energetic stress, but how AMPK activates TFEB remains unresolved. We demonstrate that AMPK directly phosphorylates five conserved serine residues in folliculin-interacting protein 1 (FNIP1), suppressing the function of the folliculin (FLCN)-FNIP1 complex. FNIP1 phosphorylation is required for AMPK to induce nuclear translocation of TFEB and TFEB-dependent increases of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and estrogen-related receptor alpha (ERRα) messenger RNAs. Thus, mitochondrial damage triggers AMPK-FNIP1-dependent nuclear translocation of TFEB, inducing sequential waves of lysosomal and mitochondrial biogenesis.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
