

Metabolism of low-density lipoproteins by cultured hepatocytes from normal and homozygous familial hypercholesterolemic subjects
- PMID: 3707989
- PMCID: PMC3006434
- DOI: 10.1016/0005-2760(86)90054-8
Metabolism of low-density lipoproteins by cultured hepatocytes from normal and homozygous familial hypercholesterolemic subjects
Abstract
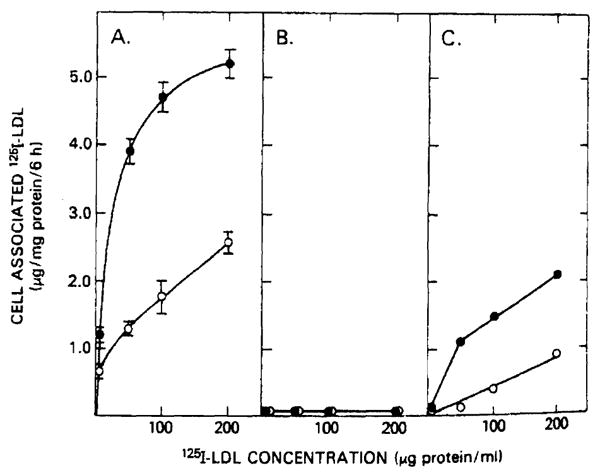
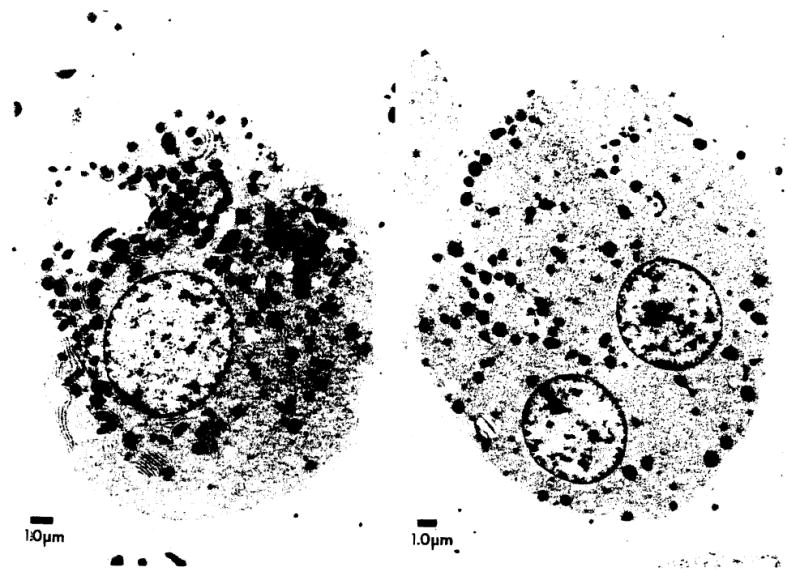
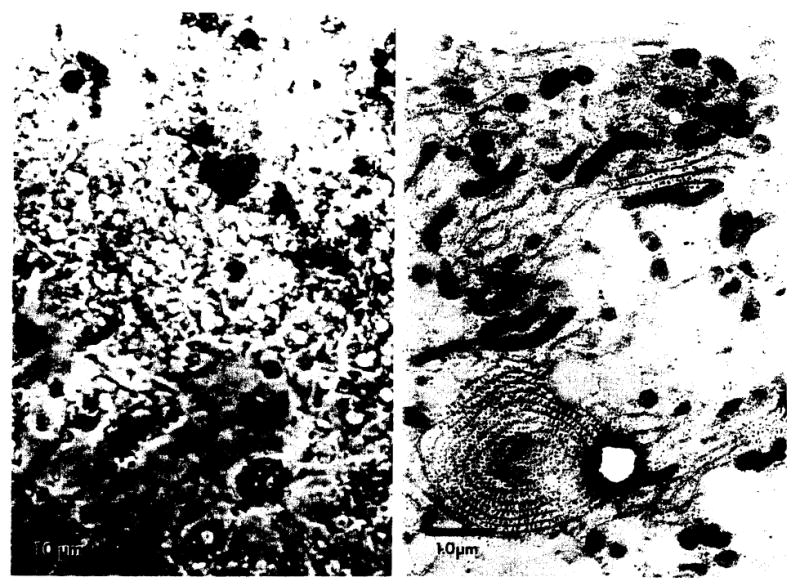
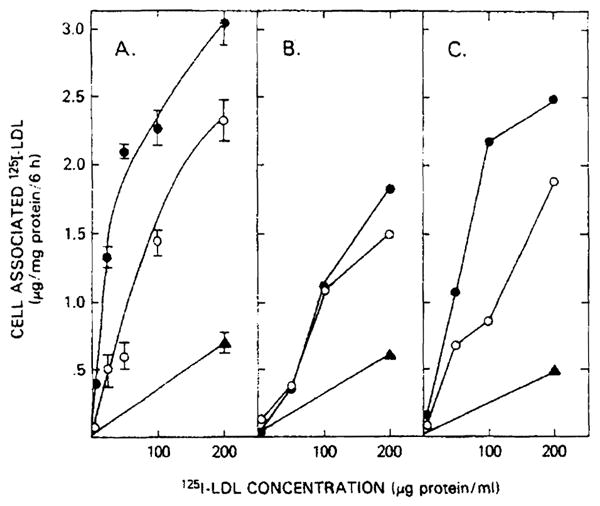
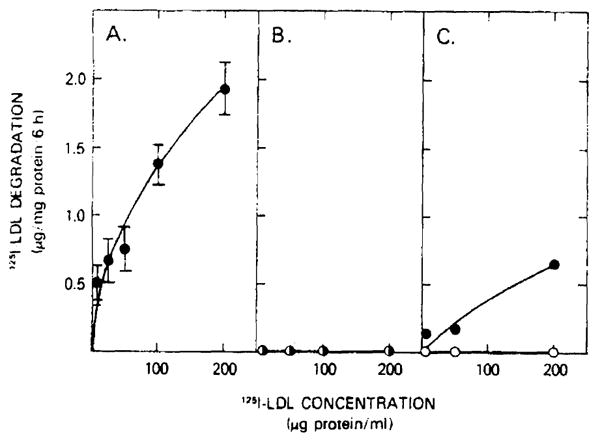
The profoundly elevated concentrations of low-density lipoproteins (LDL) present in homozygous familial hypercholesterolemia lead to symptomatic cardiovascular disease and death by early adulthood. Studies conducted in nonhepatic tissues demonstrated defective cellular recognition and metabolism of LDL in these patients. Since mammalian liver removes at least half of the LDL in the circulation, the metabolism of LDL by cultured hepatocytes isolated from familial hypercholesterolemic homozygotes was compared to hepatocytes from normal individuals. Fibroblast studies demonstrated that the familial hypercholesterolemic subjects studied were LDL receptor-negative (less than 1% normal receptor activity) and LDL receptor-defective (18% normal receptor activity). Cholesterol-depleted hepatocytes from normal subjects bound and internalized 125I-labeled LDL (Bmax = 2.2 micrograms LDL/mg cell protein). Preincubation of normal hepatocytes with 200 micrograms/ml LDL reduced binding and internalization by approx. 40%. In contrast, 125I-labeled LDL binding and internalization by receptor-negative familial hypercholesterolemic hepatocytes was unaffected by cholesterol loading and considerably lower than normal. This residual LDL uptake could not be ascribed to fluid phase endocytosis as determined by [14C]sucrose uptake. The residual LDL binding by familial hypercholesterolemia hepatocytes led to a small increase in hepatocyte cholesterol content which was relatively ineffective in reducing hepatocyte 3-hydroxy-3-methylglutaryl-CoA reductase activity. Receptor-defective familial hypercholesterolemia hepatocytes retained some degree of regulatable 125I-labeled LDL uptake, but LDL uptake did not lead to normal hepatocyte cholesterol content or 3-hydroxy-3-methylglutaryl-CoA reductase activity. These combined results indicate that the LDL receptor abnormality present in familial hypercholesterolemia fibroblasts reflects deranged hepatocyte LDL recognition and metabolism. In addition, a low-affinity, nonsaturable uptake process for LDL is present in human liver which does not efficiently modulate hepatocyte cholesterol content or synthesis.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
