

The proteomic landscape of genome-wide genetic perturbations
- PMID: 37080200
- PMCID: PMC7615649
- DOI: 10.1016/j.cell.2023.03.026
The proteomic landscape of genome-wide genetic perturbations
Abstract
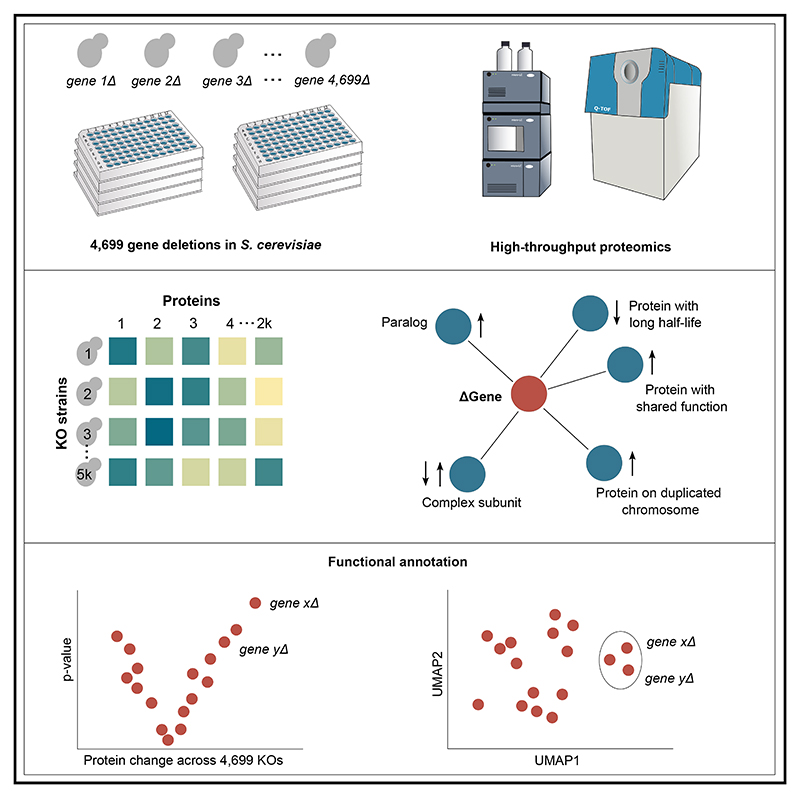
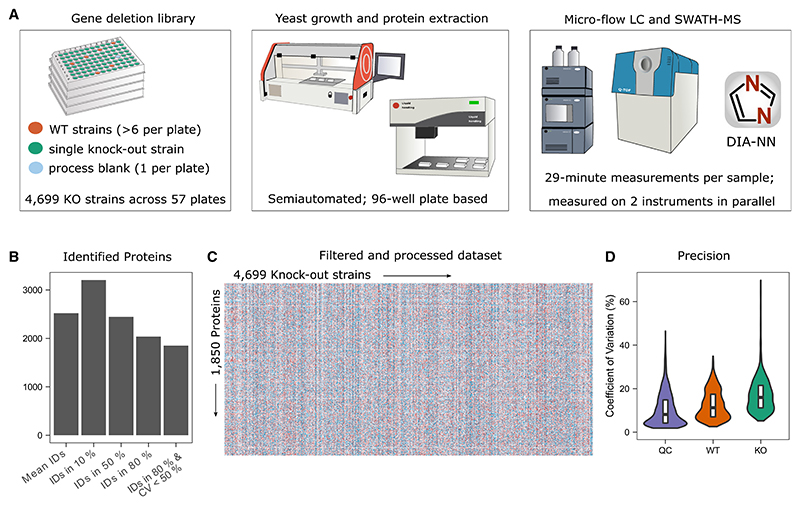
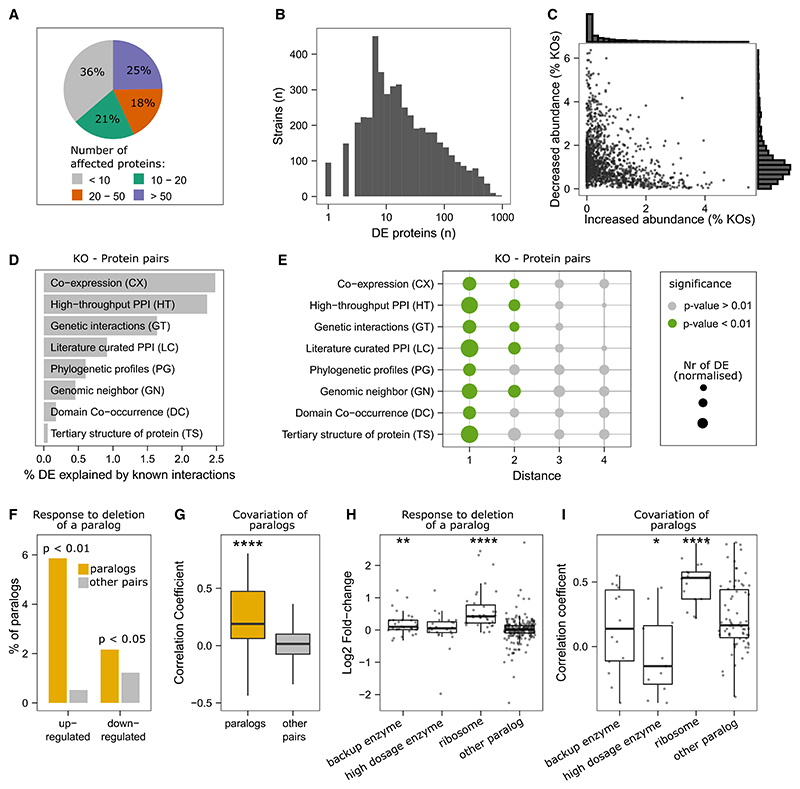
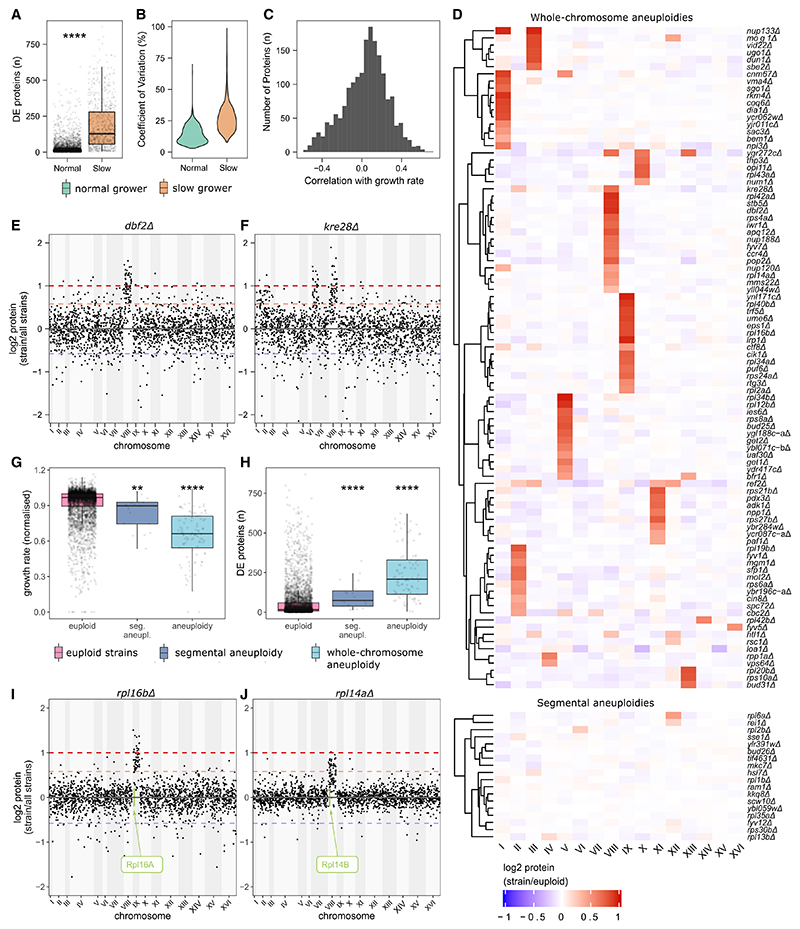
Functional genomic strategies have become fundamental for annotating gene function and regulatory networks. Here, we combined functional genomics with proteomics by quantifying protein abundances in a genome-scale knockout library in Saccharomyces cerevisiae, using data-independent acquisition mass spectrometry. We find that global protein expression is driven by a complex interplay of (1) general biological properties, including translation rate, protein turnover, the formation of protein complexes, growth rate, and genome architecture, followed by (2) functional properties, such as the connectivity of a protein in genetic, metabolic, and physical interaction networks. Moreover, we show that functional proteomics complements current gene annotation strategies through the assessment of proteome profile similarity, protein covariation, and reverse proteome profiling. Thus, our study reveals principles that govern protein expression and provides a genome-spanning resource for functional annotation.
Keywords: Saccharomyces cerevisiae; data-independent acquisition; deletion; functional genomics; functional proteomics; gene annotation; high throughput; knockout; quantitative proteomics; systems biology.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Gstaiger M, Aebersold R. Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat Rev Genet. 2009;10:617–627. - PubMed
-
- Larance M, Lamond AI. Multidimensional proteomics for cell biology. Nat Rev Mol Cell Biol. 2015;16:269–280. - PubMed
-
- Bensimon A, Heck AJ, Aebersold R. Mass spectrometry–based proteomics and network biology. Annu Rev Biochem. 2012;81:379–405. - PubMed
-
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- BB/N015282/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- IA 200829/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- FC001134/MRC_/Medical Research Council/United Kingdom
- MR/T03050X/1/MRC_/Medical Research Council/United Kingdom
- BB/N015215/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
