

Ex vivo drug response heterogeneity reveals personalized therapeutic strategies for patients with multiple myeloma
- PMID: 37081258
- PMCID: PMC10212768
- DOI: 10.1038/s43018-023-00544-9
Ex vivo drug response heterogeneity reveals personalized therapeutic strategies for patients with multiple myeloma
Abstract
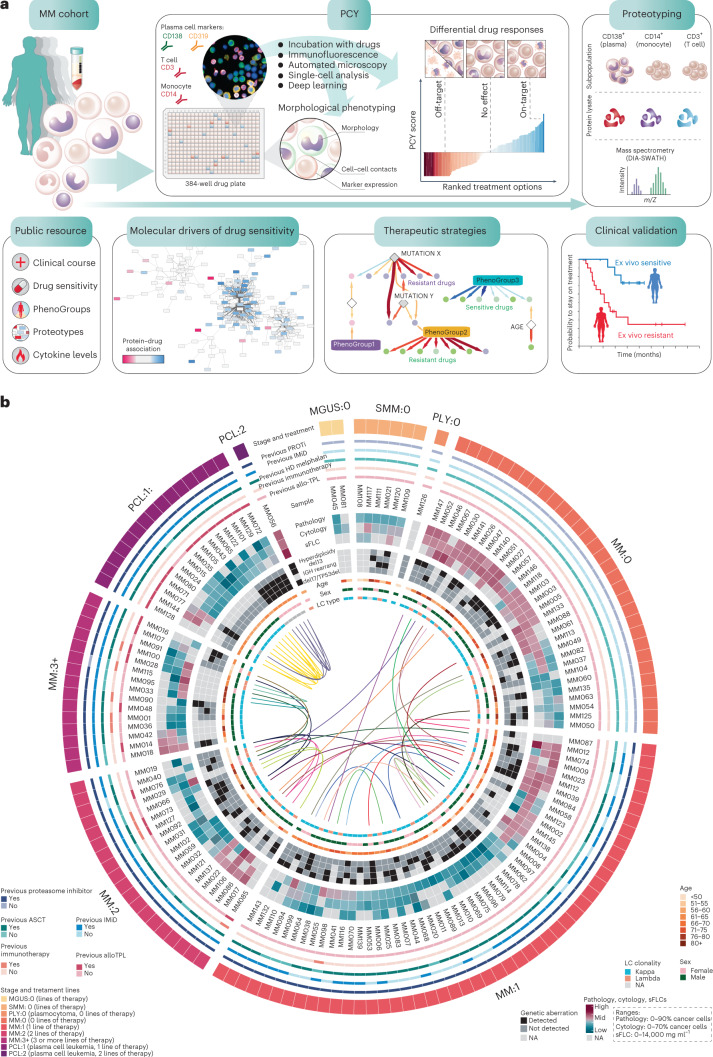
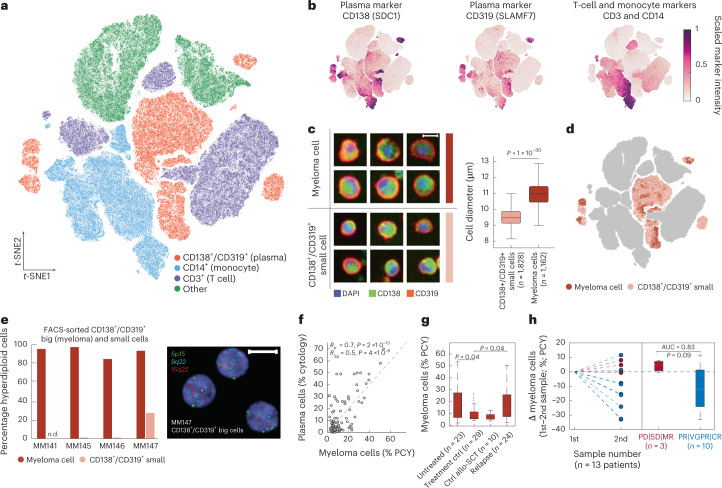
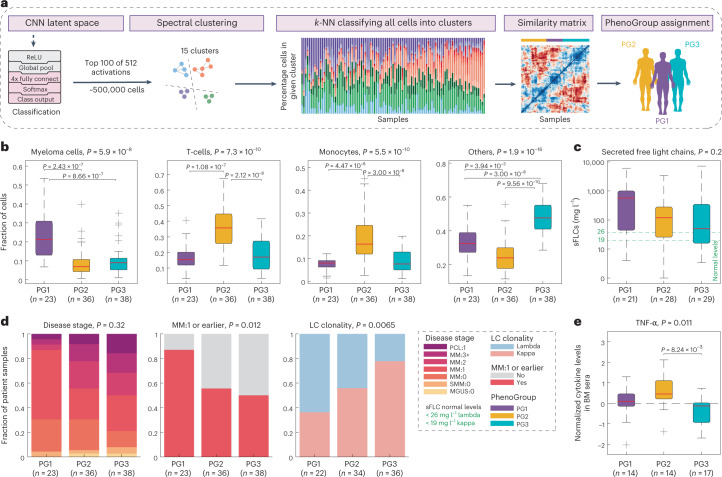
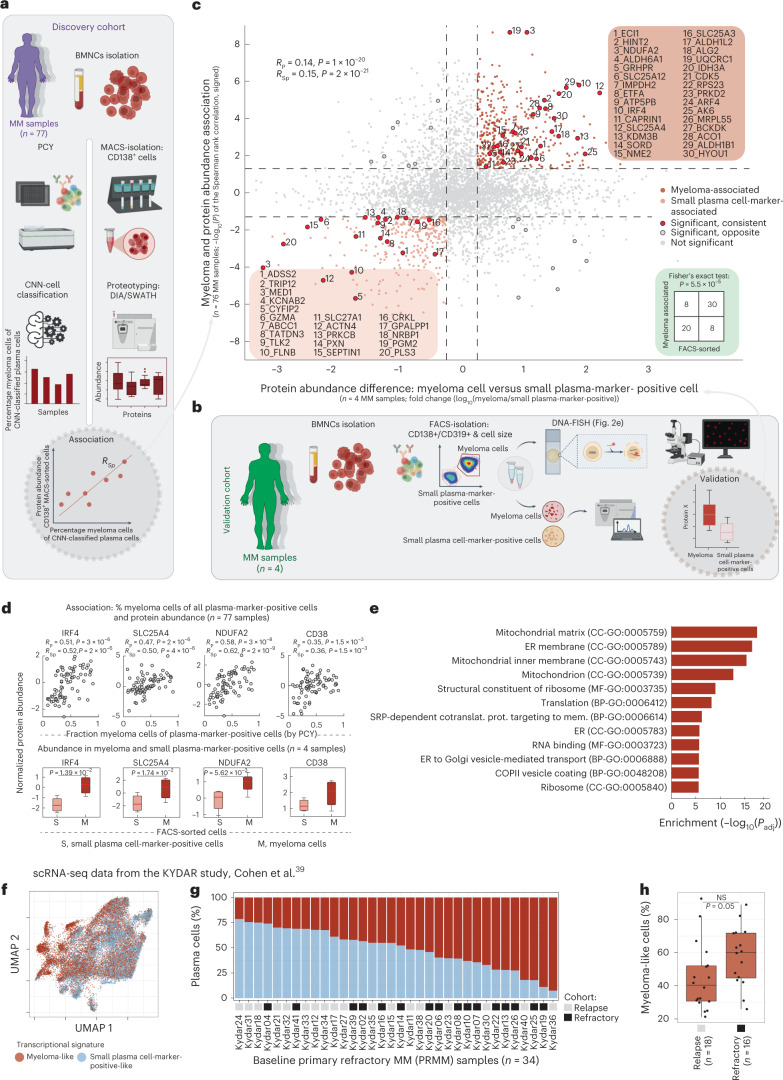
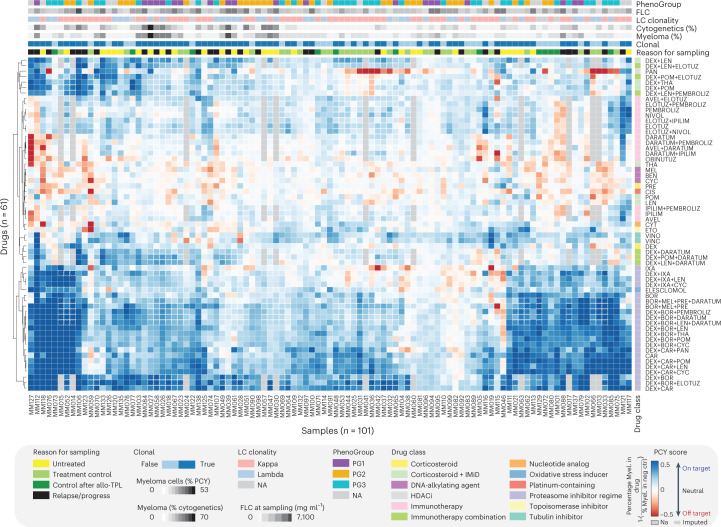
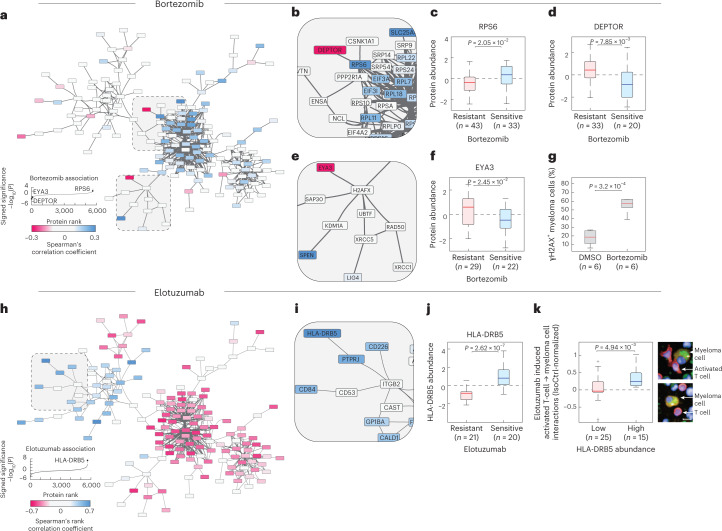
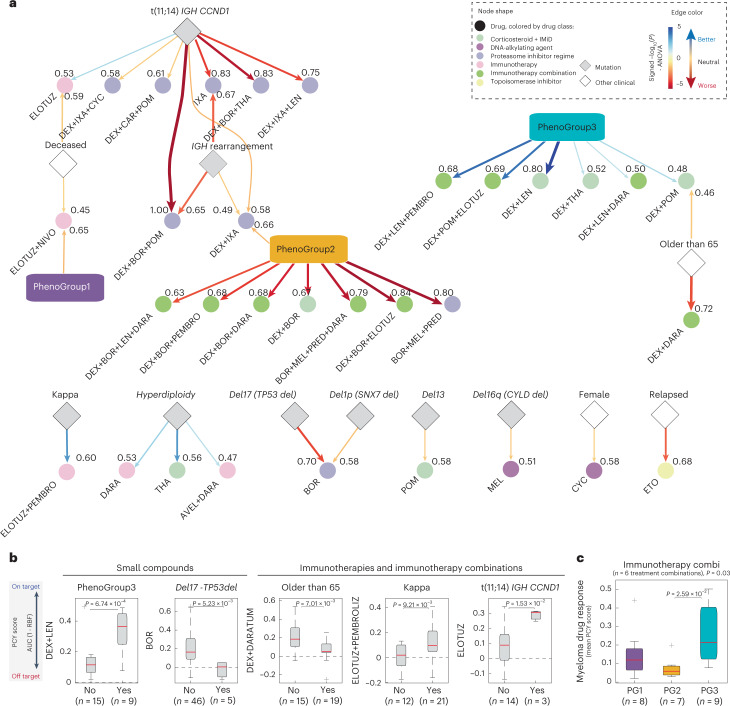
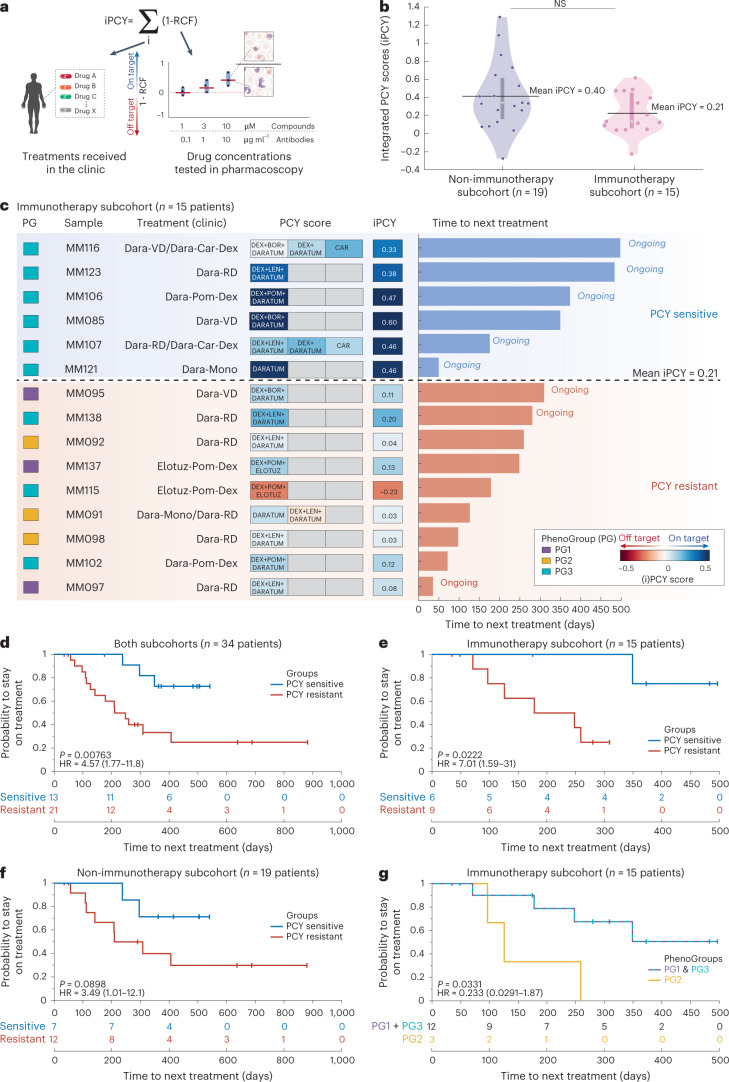
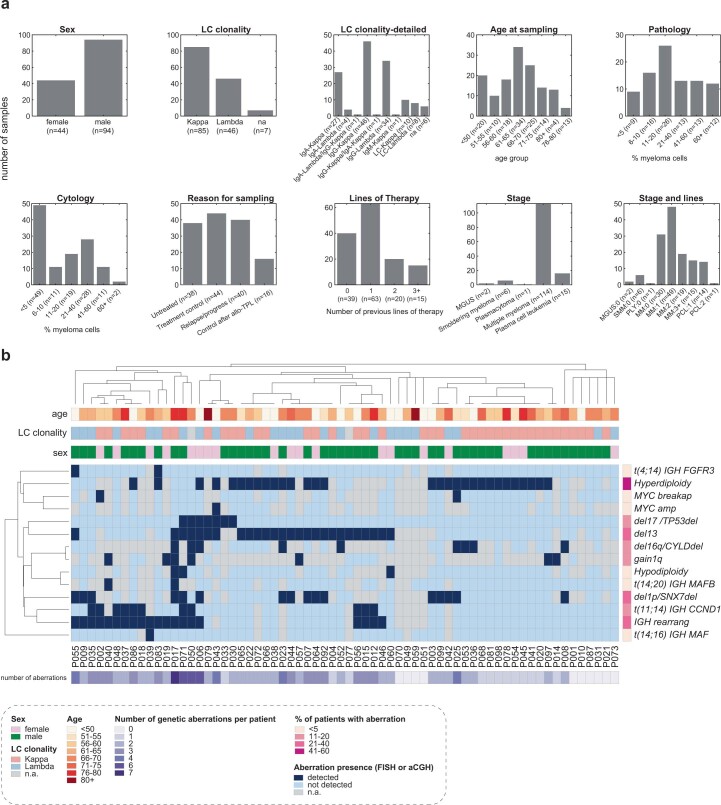
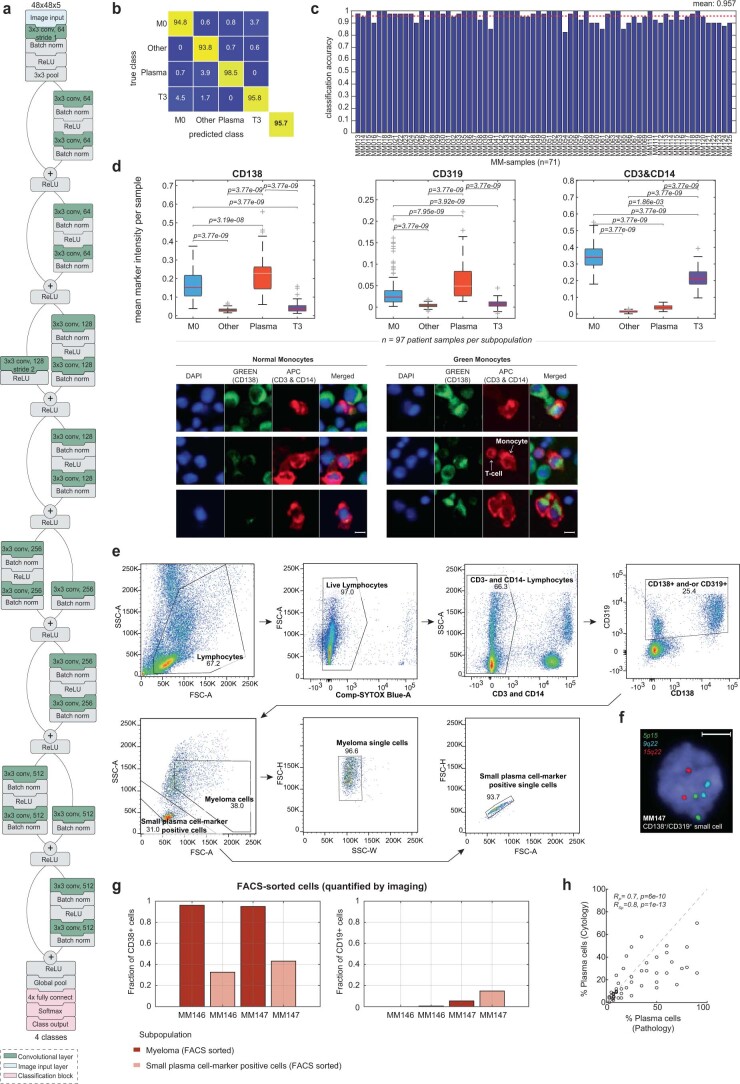
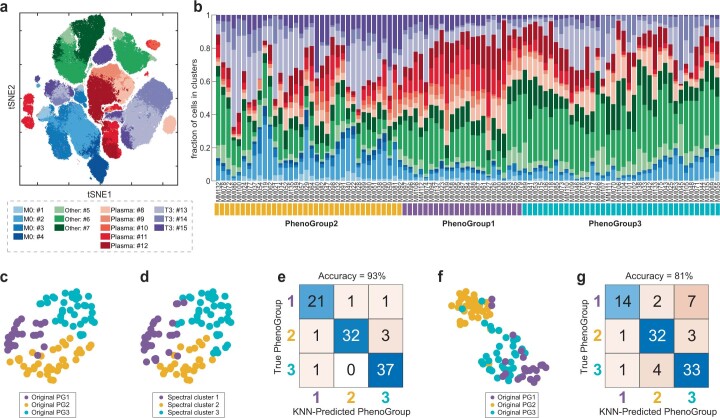
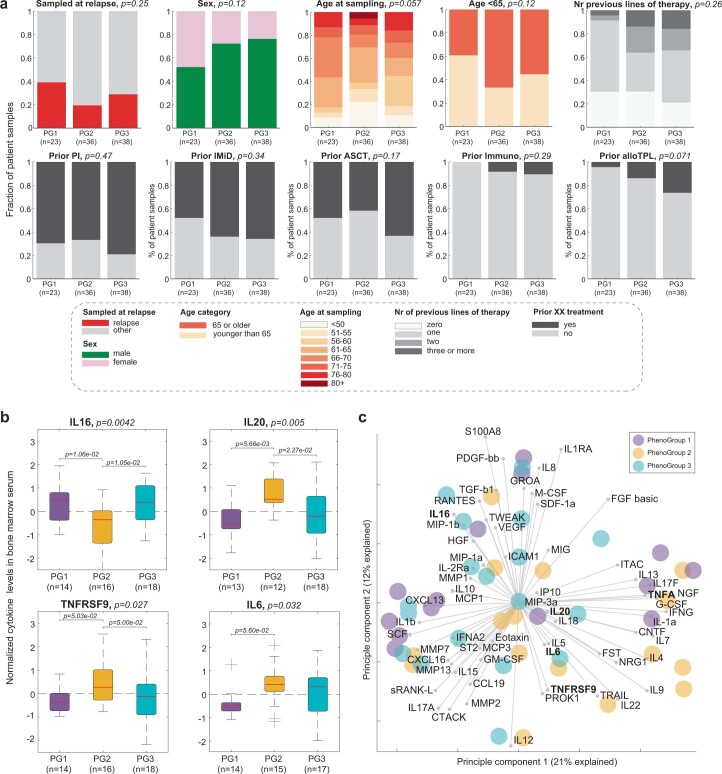
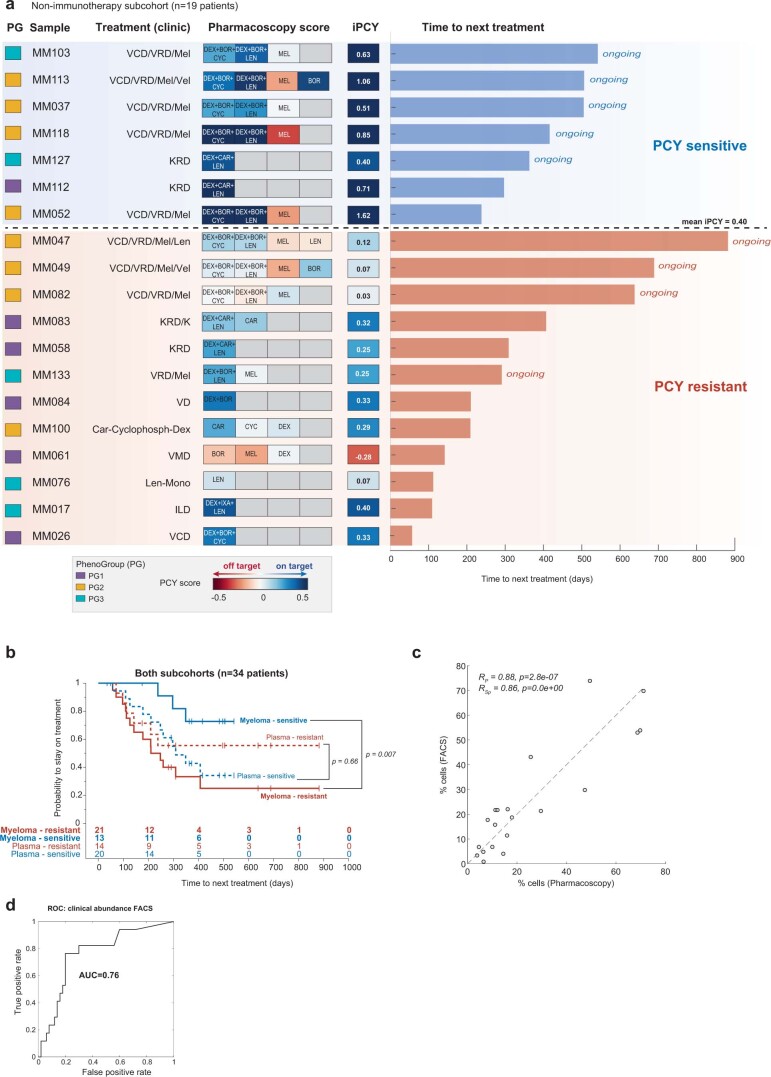
Multiple myeloma (MM) is a plasma cell malignancy defined by complex genetics and extensive patient heterogeneity. Despite a growing arsenal of approved therapies, MM remains incurable and in need of guidelines to identify effective personalized treatments. Here, we survey the ex vivo drug and immunotherapy sensitivities across 101 bone marrow samples from 70 patients with MM using multiplexed immunofluorescence, automated microscopy and deep-learning-based single-cell phenotyping. Combined with sample-matched genetics, proteotyping and cytokine profiling, we map the molecular regulatory network of drug sensitivity, implicating the DNA repair pathway and EYA3 expression in proteasome inhibitor sensitivity and major histocompatibility complex class II expression in the response to elotuzumab. Globally, ex vivo drug sensitivity associated with bone marrow microenvironmental signatures reflecting treatment stage, clonality and inflammation. Furthermore, ex vivo drug sensitivity significantly stratified clinical treatment responses, including to immunotherapy. Taken together, our study provides molecular and actionable insights into diverse treatment strategies for patients with MM.
© 2023. The Author(s).
Conflict of interest statement
B.S. was a scientific co-founder of Allcyte, which has been acquired by Exscientia. B.S. is a shareholder of Exscientia and a co-inventor on US patent application 15/514,045 relevant to the study. B.S. declares research funding from Roche and speaker fees from Novartis, GSK and AbbVie. All other authors declare no competing interests.
Figures
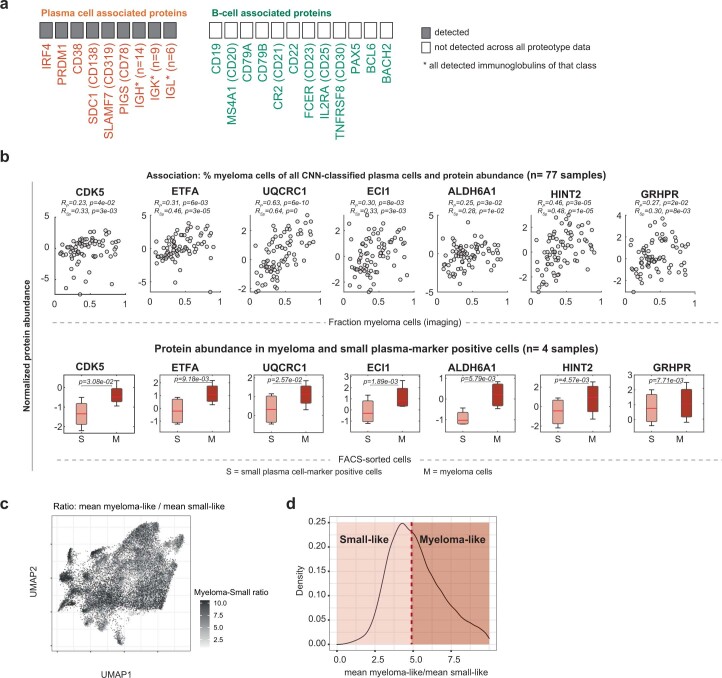
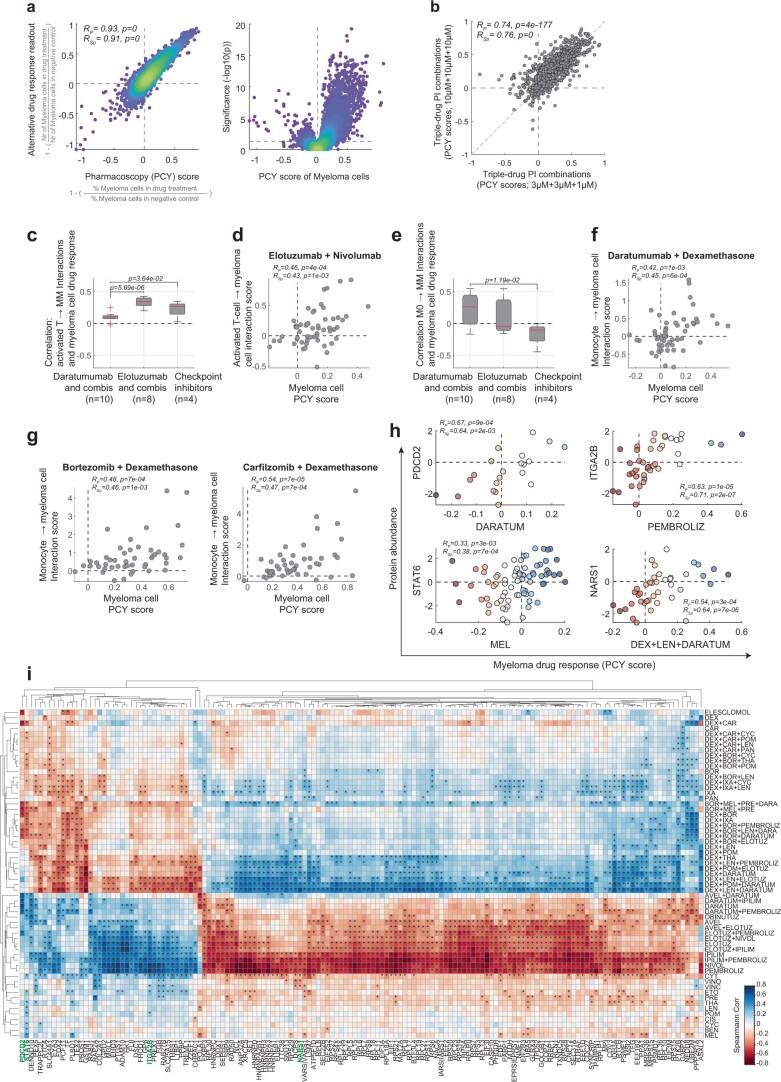
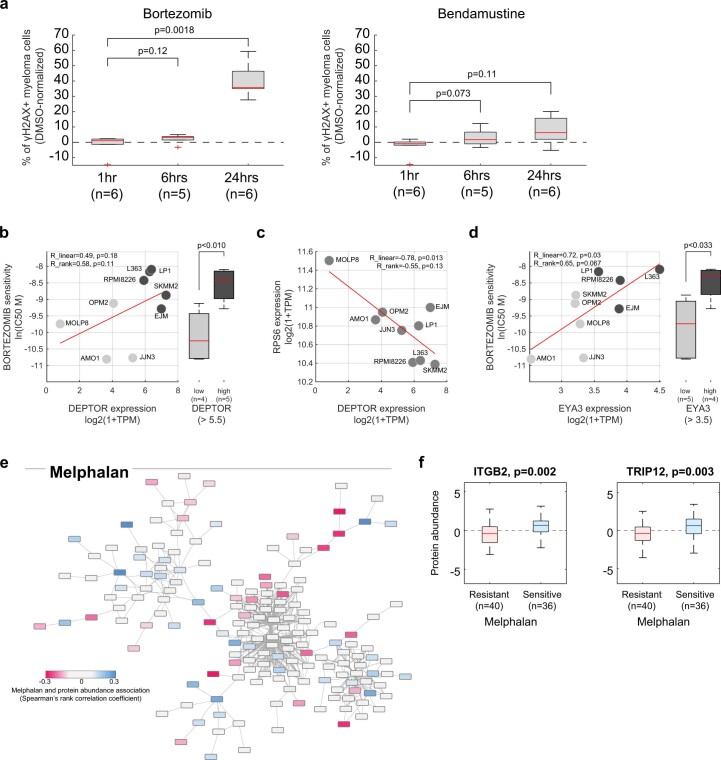
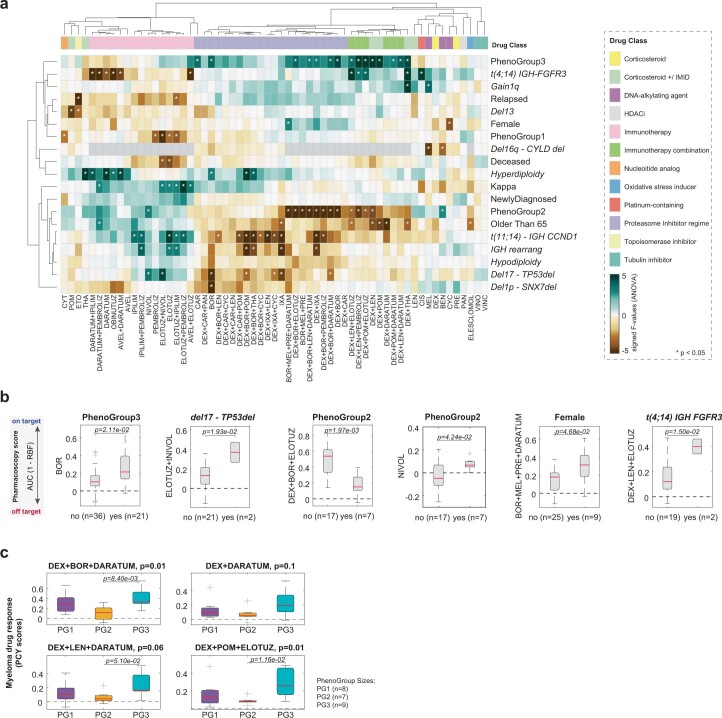
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
