

Current clinical understanding and effectiveness of portopulmonary hypertension treatment
- PMID: 37081835
- PMCID: PMC10110923
- DOI: 10.3389/fmed.2023.1142836
Current clinical understanding and effectiveness of portopulmonary hypertension treatment
Abstract
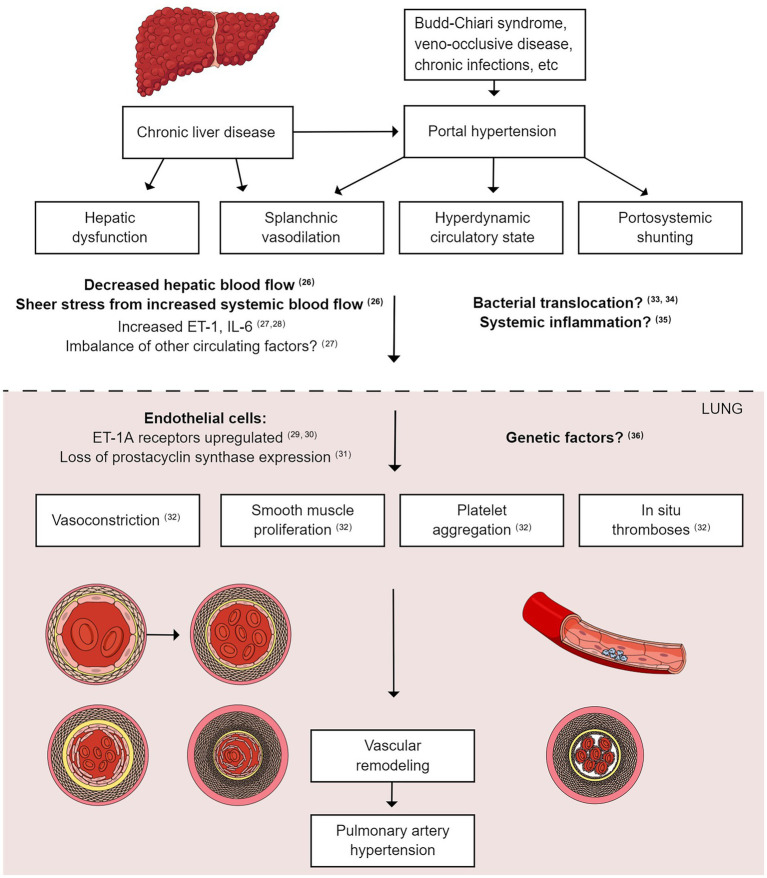
Portopulmonary hypertension (PoPH) is a rare subtype of Group 1 pulmonary arterial hypertension (PAH) with a poor prognosis. According to the most up-to-date definition, PoPH is characterized by a mean pulmonary arterial pressure (PAP) of >20 mmHg at rest, a pulmonary artery wedge pressure of ≤15 mmHg, and a pulmonary vascular resistance (PVR) of >2 Wood units with portal hypertension. Like PAH, PoPH is underpinned by an imbalance in vasoactive substances. Therefore, current guidelines recommend PAH-specific therapies for PoPH treatment; however, descriptions of the actual treatment approaches are inconsistent. Given the small patient population, PoPH is often studied in combination with idiopathic PAH; however, recent evidence suggests important differences between PoPH and idiopathic PAH in terms of hemodynamic parameters, treatment approaches, survival, socioeconomic status, and healthcare utilization. Therefore, large, multi-center registry studies are needed to examine PoPH in isolation while obtaining statistically meaningful results. PoPH has conventionally been excluded from clinical drug trials because of concerns over hepatotoxicity. Nevertheless, newer-generation endothelin receptor antagonists have shown great promise in the treatment of PoPH, reducing PVR, PAP, and World Health Organization functional class without causing hepatotoxicity. The role of liver transplantation as a treatment option for PoPH has also been controversial; however, recent evidence shows that this procedure may be beneficial in this patient population. In the future, given the shortage of liver donors, predictors of a favorable response to liver transplantation should be determined to select the most eligible patients. Collectively, advances in these three areas could help to standardize PoPH treatment in the clinic.
Keywords: endothelin receptor antagonist; liver transplantation; portopulmonary hypertension; pulmonary arterial hypertension; screening; treatment.
Copyright © 2023 Tamura, Tamura, Taniguchi and Atsukawa.
Conflict of interest statement
YuiT has received remuneration from Janssen Pharmaceuticals and Daiichi Sankyo, as well as research funds from Mochida. YT has received research grants from Janssen Pharmaceuticals and Nippon Shinyaku. MA has received remuneration from Janssen Pharmaceuticals.The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffrey CS, et al. . Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation. (2010) 122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials