

Origin of the Phosphoprotein Phosphatase (PPP) sequence family in Bacteria: Critical ancestral sequence changes, radiation patterns and substrate binding features
- PMID: 37082010
- PMCID: PMC10074919
- DOI: 10.1016/j.bbadva.2021.100005
Origin of the Phosphoprotein Phosphatase (PPP) sequence family in Bacteria: Critical ancestral sequence changes, radiation patterns and substrate binding features
Abstract
Background: Phosphoprotein phosphatases (PPP) belong to the PPP Sequence family, which in turn belongs to the broader metallophosphoesterase (MPE) superfamily. The relationship between the PPP Sequence family and other members of the MPE superfamily remains unresolved, in particular what transitions took place in an ancestral MPE to ultimately produce the phosphoprotein specific phosphatases (PPPs).
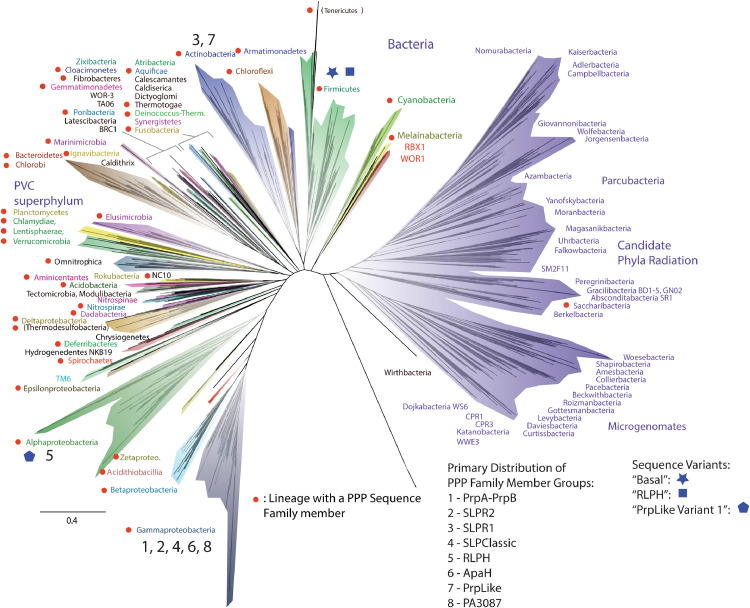
Methods: We use structural and sequence alignment data, phylogenetic tree analysis, sequence signature (Weblogo) analysis, in silico protein-peptide modeling data, and in silico mutagenesis to trace a likely route of evolution from MPEs to the PPP Sequence family. Hidden Markov Model (HMM) based iterative database search strategies were utilized to identify PPP Sequence Family members from numerous bacterial groups.
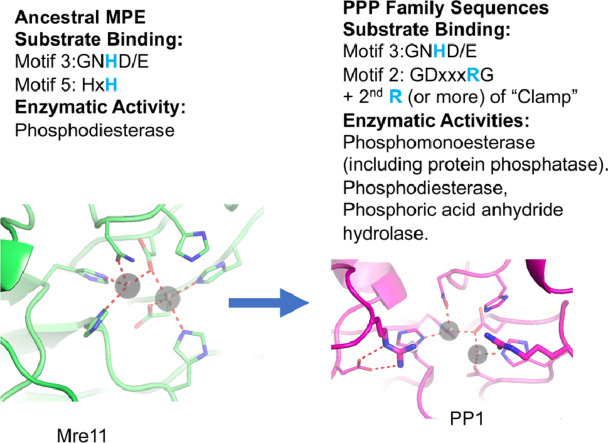
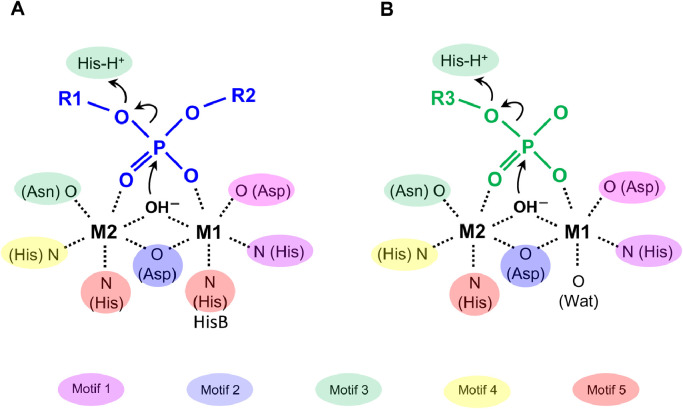
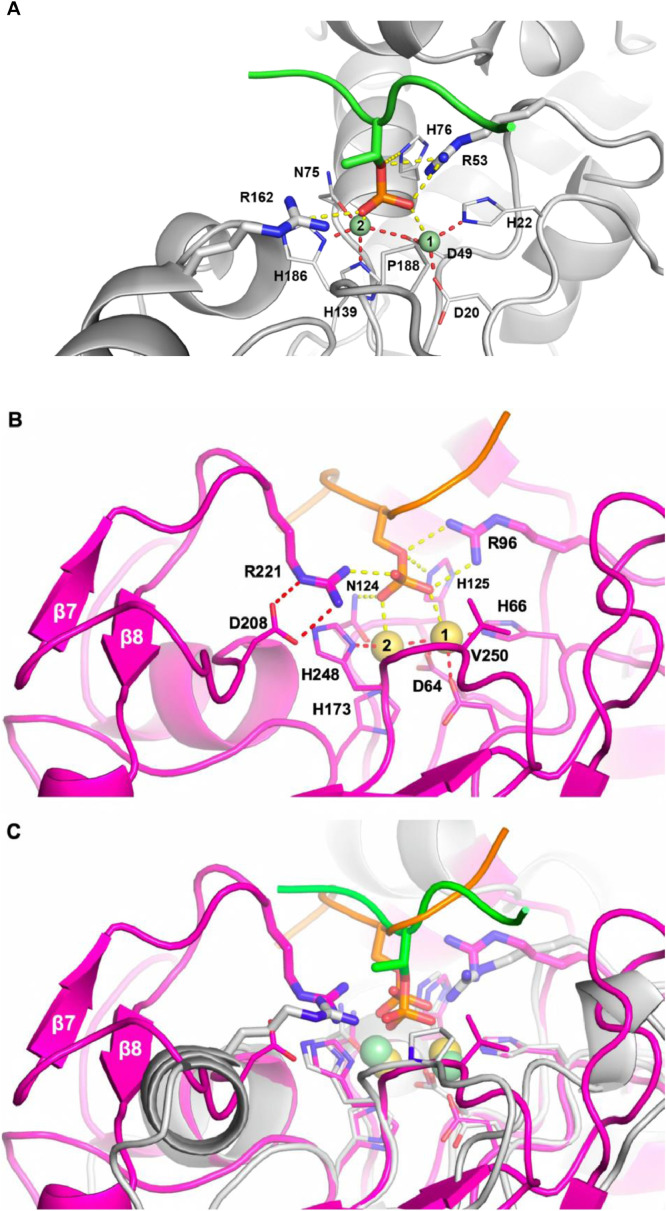
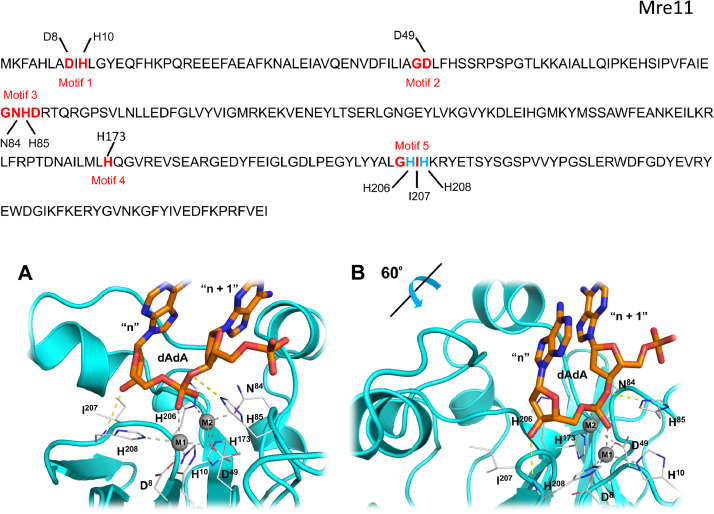
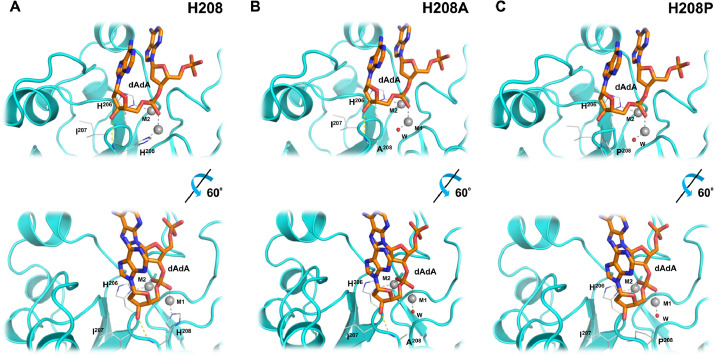
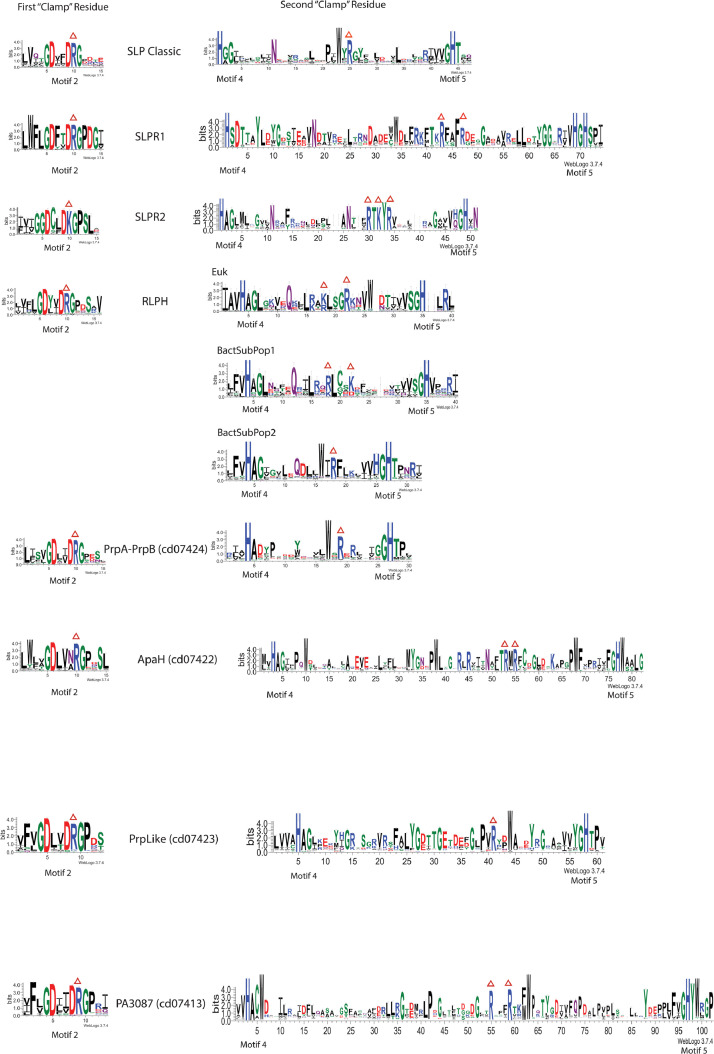
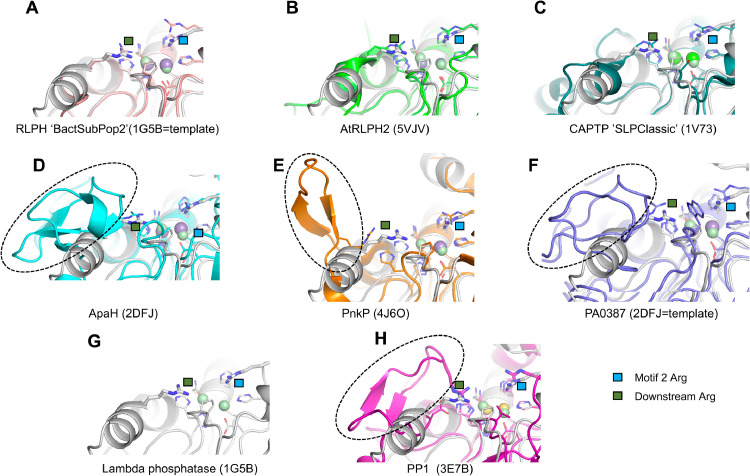
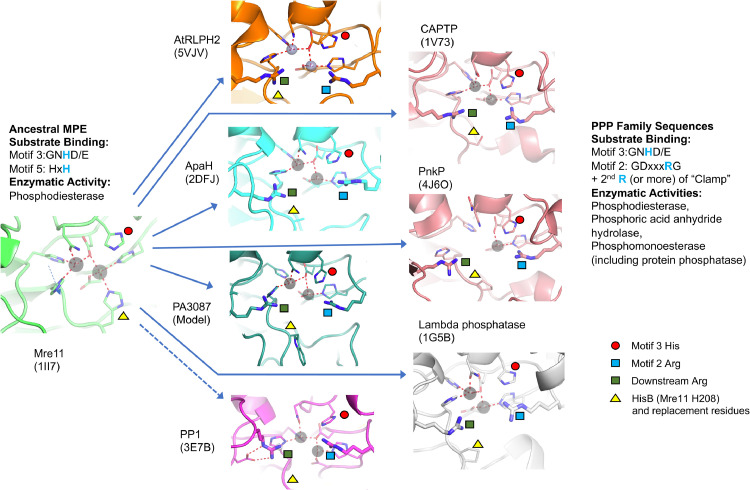
Results: Using Mre11 as proxy for an ancestral nuclease-like MPE we trace a possible evolutionary route that alters a single active site substrate binding His-residue to yield a new substrate binding accessory, the "2-Arg-Clamp". The 2-Arg-Clamp is not found in MPEs, but is present in all PPP Sequence family members, where the phosphomonesterase reaction predominates. Variation in position of the clamp arginines and a supplemental sequence loop likely provide substrate specificity for each PPP Sequence family group.
Conclusions: Loss of a key substrate binding His-in MPEs opened the path to bind novel substrates and evolution of the 2-Arg-Clamp, a sequence change seen in both bacterial and eukaryotic phosphoprotein phosphatases.General significance: We establish a likely evolutionary route from nuclease-like MPE to PPP Sequence family enzymes, that includes the phosphoprotein phosphatases.
Keywords: Bacterial origin; Metallophosphoesterase; Molecular dynamics simulation; Phosphomonoesterase; Phylogenetic analysis; Protein phosphatase.
© 2021 The Author(s). Published by Elsevier B.V.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
The origin and radiation of the phosphoprotein phosphatase (PPP) enzymes of Eukaryotes.Sci Rep. 2021 Jul 1;11(1):13681. doi: 10.1038/s41598-021-93206-8. Sci Rep. 2021. PMID: 34211082 Free PMC article.
-
Eukaryotic-like Phosphoprotein Phosphatase (PPP) enzyme evolution: interactions with environmental toxins and regulatory proteins.Biosci Rep. 2023 May 5;43(5):BSR20230378. doi: 10.1042/BSR20230378. Online ahead of print. Biosci Rep. 2023. PMID: 37144562 Free PMC article.
-
Widespread presence of "bacterial-like" PPP phosphatases in eukaryotes.BMC Evol Biol. 2004 Nov 19;4:47. doi: 10.1186/1471-2148-4-47. BMC Evol Biol. 2004. PMID: 15555063 Free PMC article.
-
The phosphoprotein phosphatase family of Ser/Thr phosphatases as principal targets of naturally occurring toxins.Crit Rev Toxicol. 2011 Feb;41(2):83-110. doi: 10.3109/10408444.2010.515564. Crit Rev Toxicol. 2011. PMID: 21288162 Review.
-
Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates.Annu Rev Biochem. 2018 Jun 20;87:921-964. doi: 10.1146/annurev-biochem-062917-012332. Annu Rev Biochem. 2018. PMID: 29925267 Review.
Cited by
-
The origin and radiation of the phosphoprotein phosphatase (PPP) enzymes of Eukaryotes.Sci Rep. 2021 Jul 1;11(1):13681. doi: 10.1038/s41598-021-93206-8. Sci Rep. 2021. PMID: 34211082 Free PMC article.
-
Plant acetyl-CoA carboxylase: The homomeric form and the heteromeric form.BBA Adv. 2025 Feb 18;7:100148. doi: 10.1016/j.bbadva.2025.100148. eCollection 2025. BBA Adv. 2025. PMID: 40060359 Free PMC article.
-
Eukaryotic-like Phosphoprotein Phosphatase (PPP) enzyme evolution: interactions with environmental toxins and regulatory proteins.Biosci Rep. 2023 May 5;43(5):BSR20230378. doi: 10.1042/BSR20230378. Online ahead of print. Biosci Rep. 2023. PMID: 37144562 Free PMC article.
References
-
- Cohen P. The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem Sci. 2000;25(12):596–601. - PubMed
-
- Sharma K., D'Souza R.C., Tyanova S., Schaab C., Wisniewski J.R., Cox J., Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8(5):1583–1594. - PubMed
-
- Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. - PubMed
-
- Chen M.J., Dixon J.E., Manning G. Genomics and evolution of protein phosphatases. Sci Signal. 2017;10(474) - PubMed
LinkOut - more resources
Full Text Sources
