

Modeling of Ca2+ transients initiated by GPCR agonists in mesenchymal stromal cells
- PMID: 37082025
- PMCID: PMC10074909
- DOI: 10.1016/j.bbadva.2021.100012
Modeling of Ca2+ transients initiated by GPCR agonists in mesenchymal stromal cells
Abstract
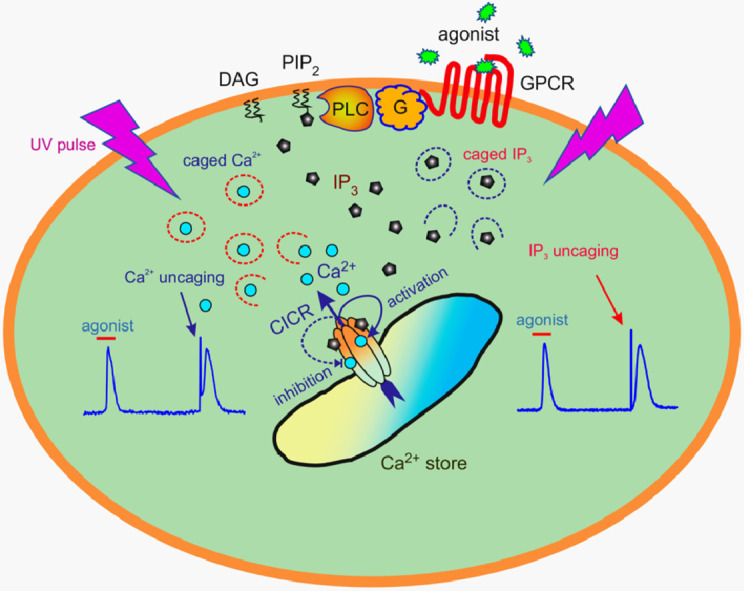
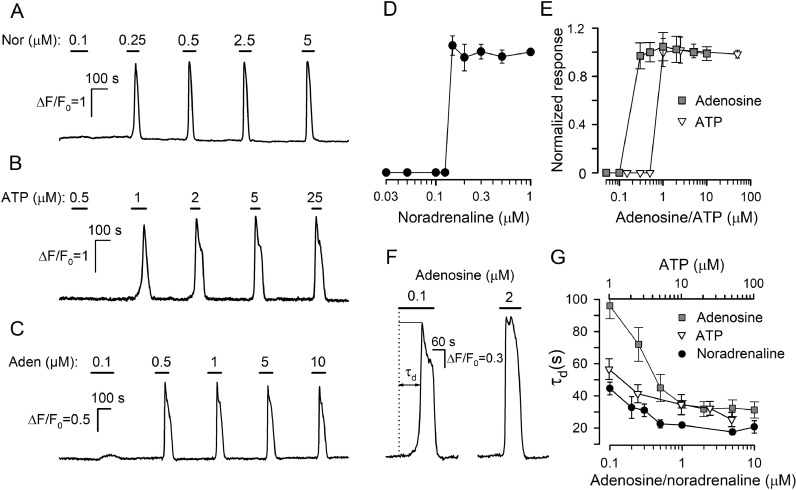
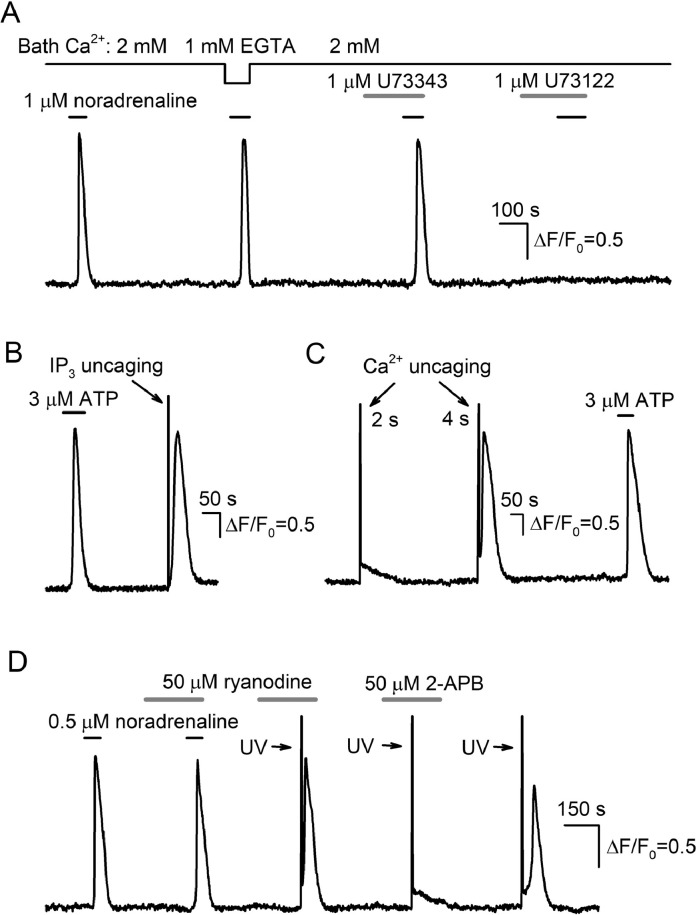
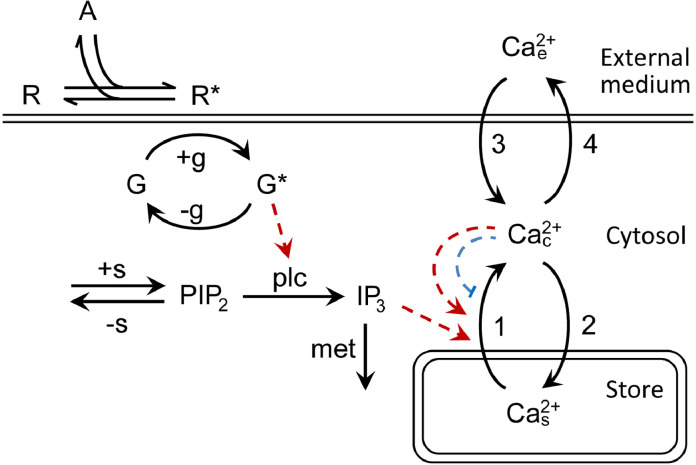
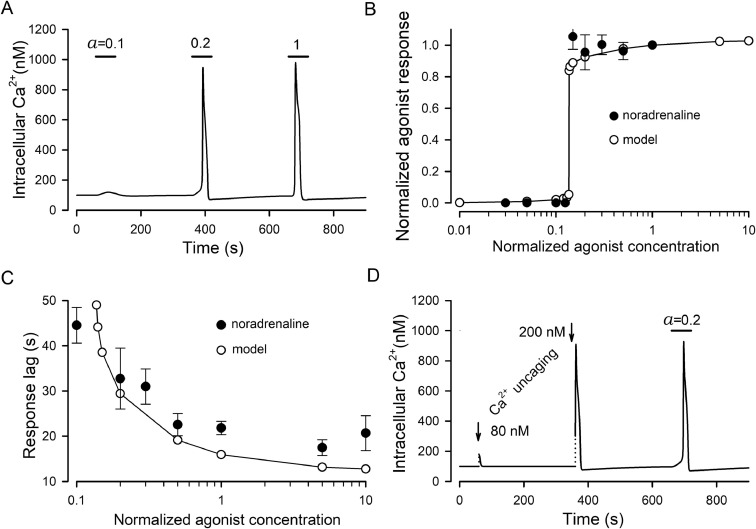
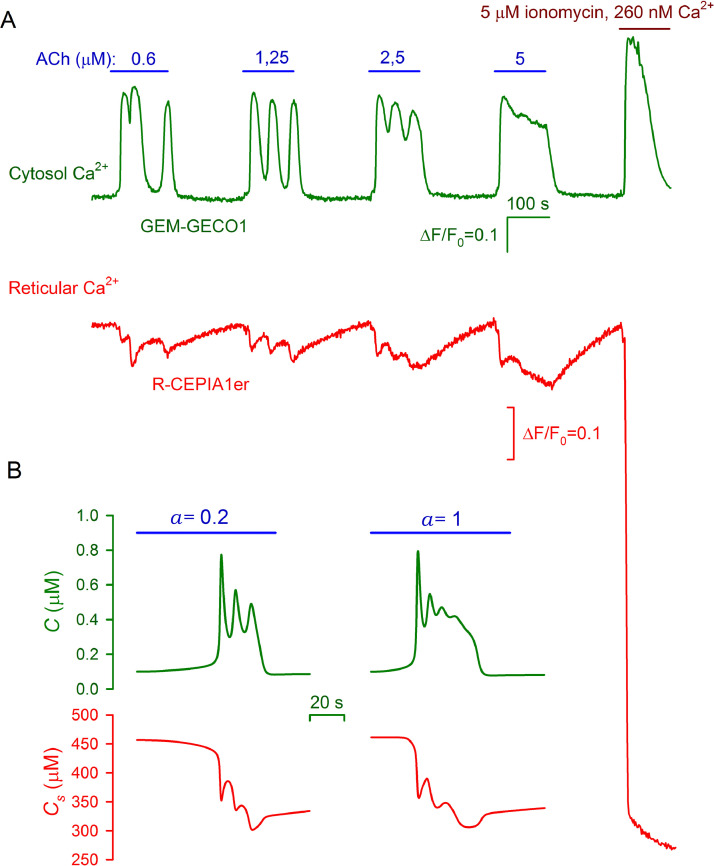
The integrative study that included experimentation and mathematical modeling was carried out to analyze dynamic aspects of transient Ca2+ signaling induced by brief pulses of GPCR agonists in mesenchymal stromal cells from the human adipose tissue (AD-MSCs). The experimental findings argued for IP3/Ca2+-regulated Ca2+ release via IP3 receptors (IP3Rs) as a key mechanism mediating agonist-dependent Ca2+ transients. The consistent signaling circuit was proposed to formalize coupling of agonist binding to Ca2+ mobilization for mathematical modeling. The model properly simulated the basic phenomenology of agonist transduction in AD-MSCs, which mostly produced single Ca2+ spikes upon brief stimulation. The spike-like responses were almost invariantly shaped at different agonist doses above a threshold, while response lag markedly decreased with stimulus strength. In AD-MSCs, agonists and IP3 uncaging elicited similar Ca2+ transients but IP3 pulses released Ca2+ without pronounced delay. This suggested that IP3 production was rate-limiting in agonist transduction. In a subpopulation of AD-MSCs, brief agonist pulses elicited Ca2+ bursts crowned by damped oscillations. With properly adjusted parameters of IP3R inhibition by cytosolic Ca2+, the model reproduced such oscillatory Ca2+ responses as well. GEM-GECO1 and R-CEPIA1er, the genetically encoded sensors of cytosolic and reticular Ca2+, respectively, were co-expressed in HEK-293 cells that also responded to agonists in an "all-or-nothing" manner. The experimentally observed Ca2+ signals triggered by ACh in both compartments were properly simulated with the suggested signaling circuit. Thus, the performed modeling of the transduction process provides sufficient theoretical basis for deeper interpretation of experimental findings on agonist-induced Ca2+ signaling in AD-MSCs.
Keywords: Agonist-induced Ca2+ signaling; Ca2+-induced Ca2+ release; IP3 receptors; Mathematical modeling; Mesenchymal stromal cells.
© 2021 The Author(s).
Conflict of interest statement
The authors disclose no conflict of interest.
Figures


Similar articles
-
Agonist-Induced Ca2+ Signaling in HEK-293-Derived Cells Expressing a Single IP3 Receptor Isoform.Cells. 2024 Mar 22;13(7):562. doi: 10.3390/cells13070562. Cells. 2024. PMID: 38607001 Free PMC article.
-
Functional expression of adrenoreceptors in mesenchymal stromal cells derived from the human adipose tissue.Biochim Biophys Acta. 2014 Sep;1843(9):1899-908. doi: 10.1016/j.bbamcr.2014.05.002. Epub 2014 May 16. Biochim Biophys Acta. 2014. PMID: 24841820
-
Coupling of P2Y receptors to Ca2+ mobilization in mesenchymal stromal cells from the human adipose tissue.Cell Calcium. 2018 May;71:1-14. doi: 10.1016/j.ceca.2017.11.001. Epub 2017 Nov 7. Cell Calcium. 2018. PMID: 29604959
-
Calcium and IP3 dynamics in cardiac myocytes: experimental and computational perspectives and approaches.Front Pharmacol. 2014 Mar 6;5:35. doi: 10.3389/fphar.2014.00035. eCollection 2014. Front Pharmacol. 2014. PMID: 24639654 Free PMC article. Review.
-
Dynamic clustering of IP3 receptors by IP3.Biochem Soc Trans. 2012 Apr;40(2):325-30. doi: 10.1042/BST20110772. Biochem Soc Trans. 2012. PMID: 22435806 Review.
Cited by
-
Agonist-Induced Ca2+ Signaling in HEK-293-Derived Cells Expressing a Single IP3 Receptor Isoform.Cells. 2024 Mar 22;13(7):562. doi: 10.3390/cells13070562. Cells. 2024. PMID: 38607001 Free PMC article.
-
Taste Cells of the Type III Employ CASR to Maintain Steady Serotonin Exocytosis at Variable Ca2+ in the Extracellular Medium.Cells. 2022 Apr 18;11(8):1369. doi: 10.3390/cells11081369. Cells. 2022. PMID: 35456048 Free PMC article.
References
-
- Clapham D. Calcium signaling. Cell. 2007;131:1047–1058. - PubMed
-
- Berridge M.J., Bootman M.D., Roderick H.L. Calcium signaling: dynamics, homeostasis and remodeling. Nat. Rev. Mol. Cell Biol. 2003;4:517–529. - PubMed
-
- Rizzuto R., De Stefani D., Raffaello A., Mammucari C. Mitochondria as sensors and regulators of calcium signaling. Nat. Rev. Mol. Cell Biol. 2012;13:566–578. - PubMed
-
- Berridge M.J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 2016;96:1261–1296. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
