

A systems approach evaluating the impact of SARS-CoV-2 variant of concern mutations on CD8+ T cell responses
- PMID: 37082106
- PMCID: PMC10112682
- DOI: 10.1093/immadv/ltad005
A systems approach evaluating the impact of SARS-CoV-2 variant of concern mutations on CD8+ T cell responses
Abstract
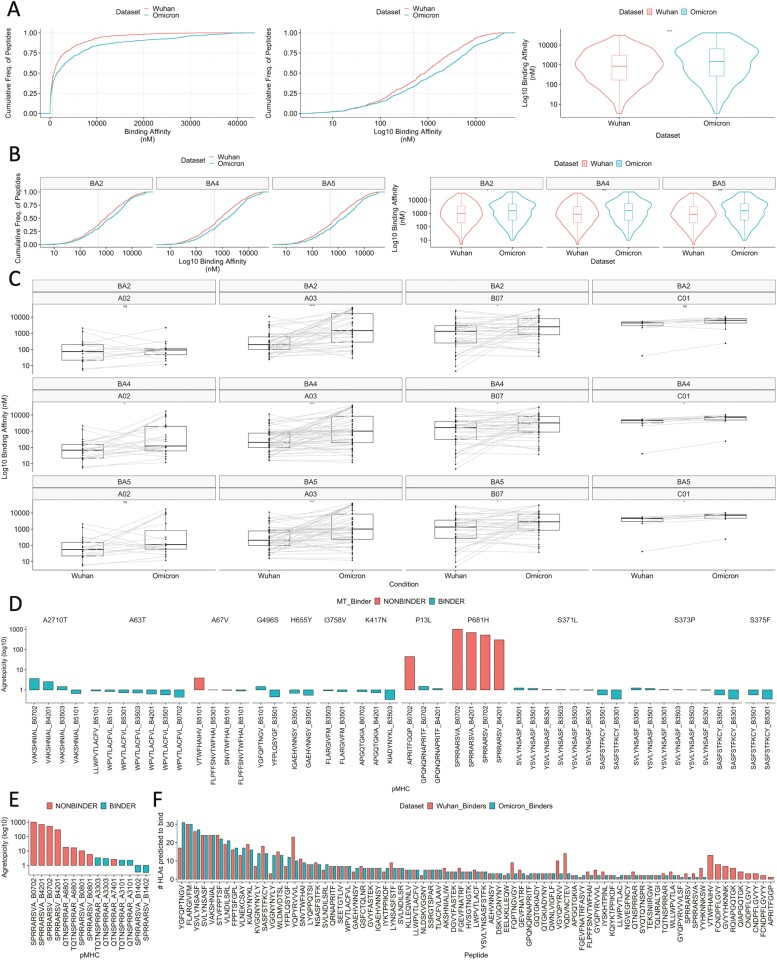
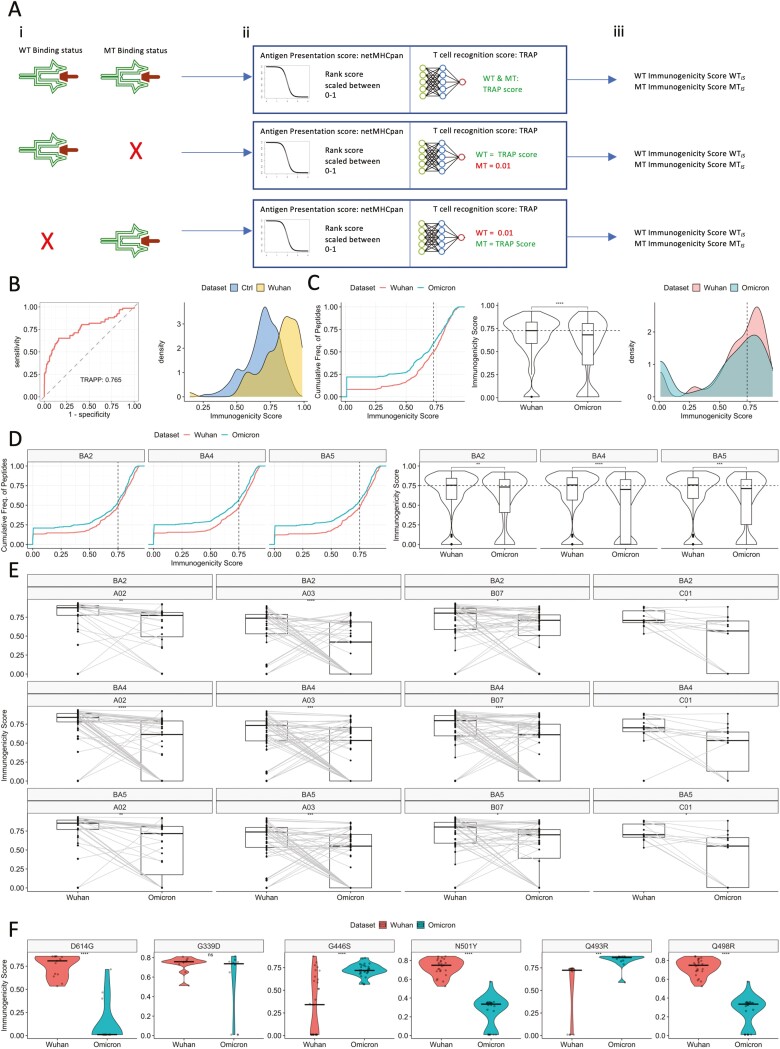
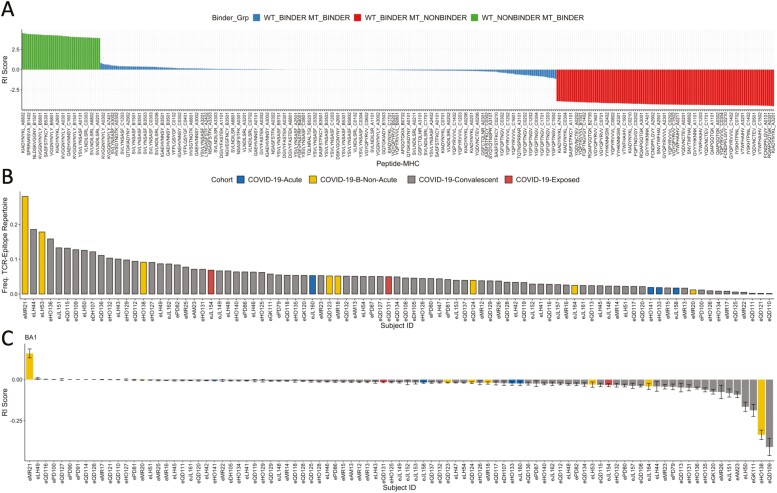
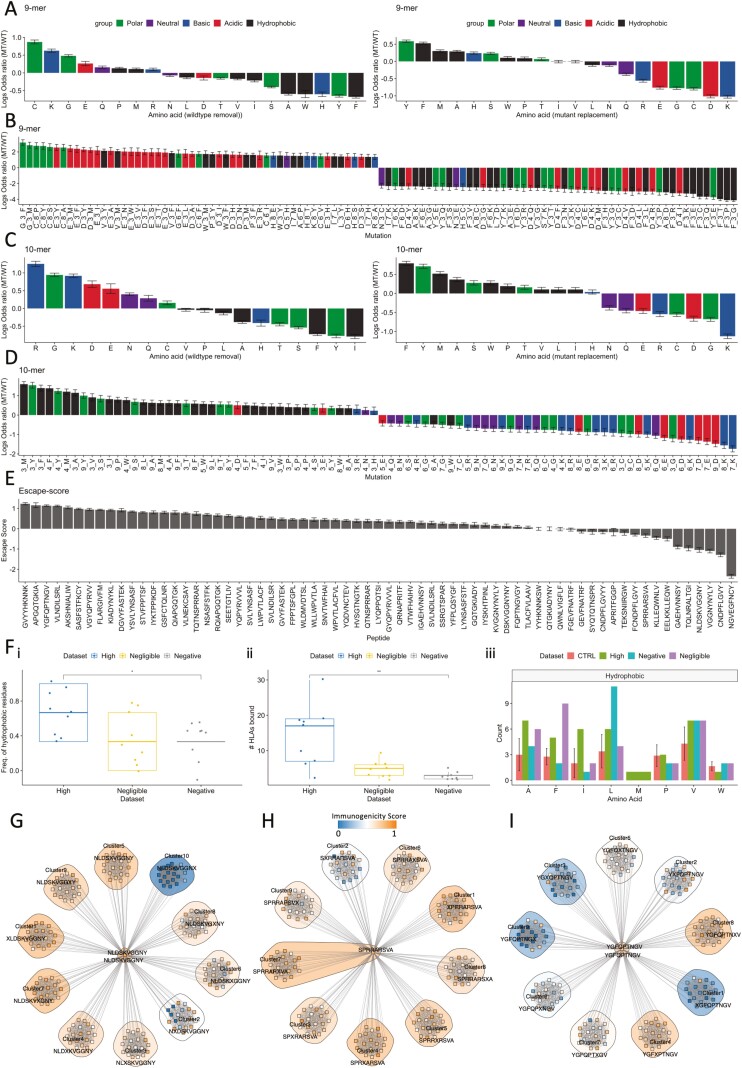
T cell recognition of SARS-CoV-2 antigens after vaccination and/or natural infection has played a central role in resolving SARS-CoV-2 infections and generating adaptive immune memory. However, the clinical impact of SARS-CoV-2-specific T cell responses is variable and the mechanisms underlying T cell interaction with target antigens are not fully understood. This is especially true given the virus' rapid evolution, which leads to new variants with immune escape capacity. In this study, we used the Omicron variant as a model organism and took a systems approach to evaluate the impact of mutations on CD8+ T cell immunogenicity. We computed an immunogenicity potential score for each SARS-CoV-2 peptide antigen from the ancestral strain and Omicron, capturing both antigen presentation and T cell recognition probabilities. By comparing ancestral vs. Omicron immunogenicity scores, we reveal a divergent and heterogeneous landscape of impact for CD8+ T cell recognition of mutated targets in Omicron variants. While T cell recognition of Omicron peptides is broadly preserved, we observed mutated peptides with deteriorated immunogenicity that may assist breakthrough infection in some individuals. We then combined our scoring scheme with an in silico mutagenesis, to characterise the position- and residue-specific theoretical mutational impact on immunogenicity. While we predict many escape trajectories from the theoretical landscape of substitutions, our study suggests that Omicron mutations in T cell epitopes did not develop under cell-mediated pressure. Our study provides a generalisable platform for fostering a deeper understanding of existing and novel variant impact on antigen-specific vaccine- and/or infection-induced T cell immunity.
Keywords: CD8 T cell response; SARS-CoV-2; T cell recognition potential; immunogenicity; machine-learning; systems immunology.
© The Author(s) 2023. Published by Oxford University Press on behalf of the British Society for Immunology.
Conflict of interest statement
H.K. provides consultancy on spatiotemporal dynamics of antigen-specific cellular immunity.
Figures
Similar articles
-
Characterizing HLA-A2-restricted CD8+ T-cell epitopes and immune responses to Omicron variants in SARS-CoV-2-inactivated vaccine recipients.Front Immunol. 2025 Mar 18;16:1534530. doi: 10.3389/fimmu.2025.1534530. eCollection 2025. Front Immunol. 2025. PMID: 40170856 Free PMC article.
-
Minimal Crossover between Mutations Associated with Omicron Variant of SARS-CoV-2 and CD8+ T-Cell Epitopes Identified in COVID-19 Convalescent Individuals.mBio. 2022 Apr 26;13(2):e0361721. doi: 10.1128/mbio.03617-21. Epub 2022 Mar 1. mBio. 2022. PMID: 35229637 Free PMC article.
-
Emerging Variants of SARS-CoV-2 and Novel Therapeutics Against Coronavirus (COVID-19).2023 May 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2023 May 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 34033342 Free Books & Documents.
-
Insight into SARS-CoV-2 Omicron variant immune escape possibility and variant independent potential therapeutic opportunities.Heliyon. 2023 Feb;9(2):e13285. doi: 10.1016/j.heliyon.2023.e13285. Epub 2023 Jan 31. Heliyon. 2023. PMID: 36744070 Free PMC article. Review.
-
The Biological Functions and Clinical Significance of SARS-CoV-2 Variants of Corcern.Front Med (Lausanne). 2022 May 20;9:849217. doi: 10.3389/fmed.2022.849217. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35669924 Free PMC article. Review.
Cited by
-
A robust deep learning workflow to predict CD8 + T-cell epitopes.Genome Med. 2023 Sep 13;15(1):70. doi: 10.1186/s13073-023-01225-z. Genome Med. 2023. PMID: 37705109 Free PMC article.
-
Can AlphaFold's breakthrough in protein structure help decode the fundamental principles of adaptive cellular immunity?Nat Methods. 2024 May;21(5):766-776. doi: 10.1038/s41592-024-02240-7. Epub 2024 Apr 23. Nat Methods. 2024. PMID: 38654083 Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous