

Utilizing evolutionary conservation to detect deleterious mutations and improve genomic prediction in cassava
- PMID: 37082510
- PMCID: PMC10112518
- DOI: 10.3389/fpls.2022.1041925
Utilizing evolutionary conservation to detect deleterious mutations and improve genomic prediction in cassava
Abstract
Introduction: Cassava (Manihot esculenta) is an annual root crop which provides the major source of calories for over half a billion people around the world. Since its domestication ~10,000 years ago, cassava has been largely clonally propagated through stem cuttings. Minimal sexual recombination has led to an accumulation of deleterious mutations made evident by heavy inbreeding depression.
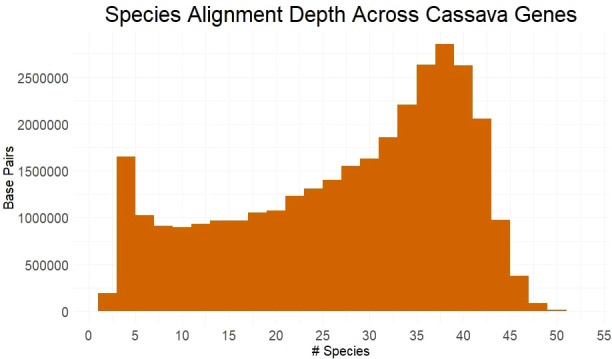
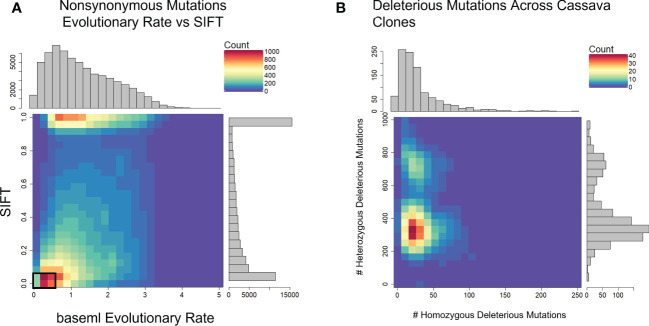
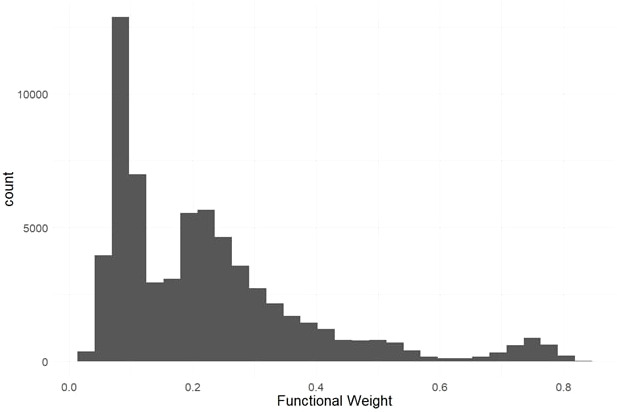
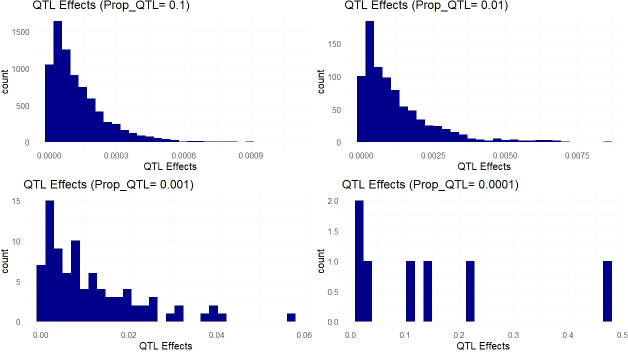
Methods: To locate and characterize these deleterious mutations, and to measure selection pressure across the cassava genome, we aligned 52 related Euphorbiaceae and other related species representing millions of years of evolution. With single base-pair resolution of genetic conservation, we used protein structure models, amino acid impact, and evolutionary conservation across the Euphorbiaceae to estimate evolutionary constraint. With known deleterious mutations, we aimed to improve genomic evaluations of plant performance through genomic prediction. We first tested this hypothesis through simulation utilizing multi-kernel GBLUP to predict simulated phenotypes across separate populations of cassava.
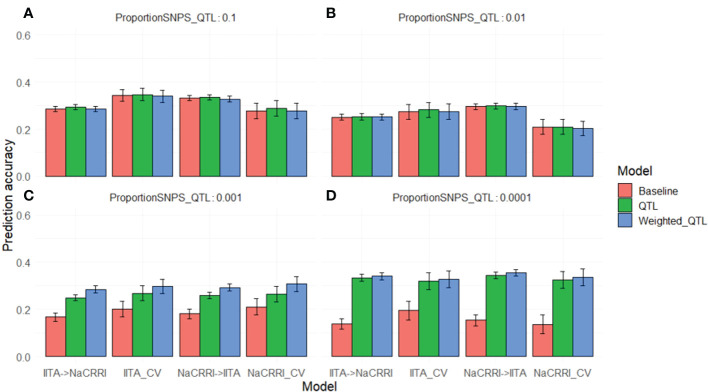
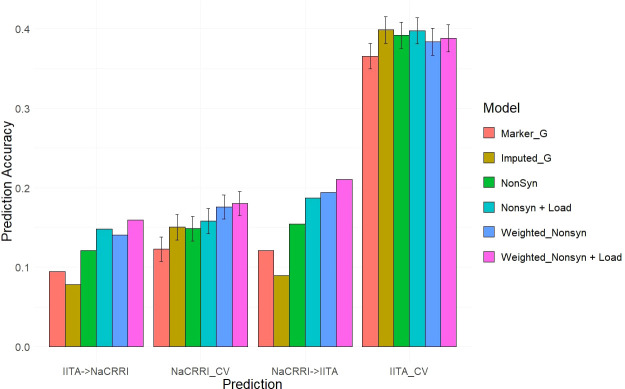
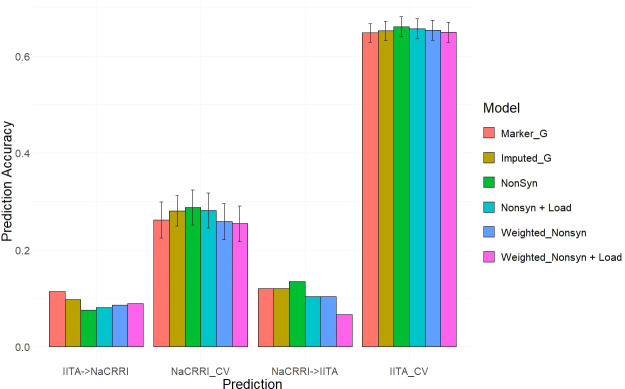
Results: Simulations showed a sizable increase of prediction accuracy when incorporating functional variants in the model when the trait was determined by<100 quantitative trait loci (QTL). Utilizing deleterious mutations and functional weights informed through evolutionary conservation, we saw improvements in genomic prediction accuracy that were dependent on trait and prediction.
Conclusion: We showed the potential for using evolutionary information to track functional variation across the genome, in order to improve whole genome trait prediction. We anticipate that continued work to improve genotype accuracy and deleterious mutation assessment will lead to improved genomic assessments of cassava clones.
Keywords: cassava (Manihot esculenta); deleterious mutation; evolutionary conservation; genetic load; genomic prediction.
Copyright © 2023 Long, Romay, Ramstein, Buckler and Robbins.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Evolutionary signatures of the erosion of sexual reproduction genes in domesticated cassava (Manihot esculenta).G3 (Bethesda). 2025 Feb 5;15(2):jkae282. doi: 10.1093/g3journal/jkae282. G3 (Bethesda). 2025. PMID: 39673428 Free PMC article.
-
Historical Introgressions from a Wild Relative of Modern Cassava Improved Important Traits and May Be Under Balancing Selection.Genetics. 2019 Dec;213(4):1237-1253. doi: 10.1534/genetics.119.302757. Epub 2019 Oct 17. Genetics. 2019. PMID: 31624088 Free PMC article.
-
Marker-Based Estimates Reveal Significant Nonadditive Effects in Clonally Propagated Cassava (Manihot esculenta): Implications for the Prediction of Total Genetic Value and the Selection of Varieties.G3 (Bethesda). 2016 Nov 8;6(11):3497-3506. doi: 10.1534/g3.116.033332. G3 (Bethesda). 2016. PMID: 27587297 Free PMC article.
-
Current knowledge and future research perspectives on cassava (Manihot esculenta Crantz) chemical defenses: An agroecological view.Phytochemistry. 2016 Oct;130:10-21. doi: 10.1016/j.phytochem.2016.05.013. Epub 2016 Jun 14. Phytochemistry. 2016. PMID: 27316676 Review.
-
The Cassava Source-Sink project: opportunities and challenges for crop improvement by metabolic engineering.Plant J. 2020 Aug;103(5):1655-1665. doi: 10.1111/tpj.14865. Epub 2020 Jun 26. Plant J. 2020. PMID: 32502321 Review.
Cited by
-
A foundational large language model for edible plant genomes.Commun Biol. 2024 Jul 9;7(1):835. doi: 10.1038/s42003-024-06465-2. Commun Biol. 2024. PMID: 38982288 Free PMC article.
-
Evolution of linear triterpenoid biosynthesis within the Euphorbia genus.Nat Commun. 2025 Jul 1;16(1):5602. doi: 10.1038/s41467-025-60708-2. Nat Commun. 2025. PMID: 40595524 Free PMC article.
-
Evolutionary signatures of the erosion of sexual reproduction genes in domesticated cassava (Manihot esculenta).G3 (Bethesda). 2025 Feb 5;15(2):jkae282. doi: 10.1093/g3journal/jkae282. G3 (Bethesda). 2025. PMID: 39673428 Free PMC article.
-
A Penalized Regression Method for Genomic Prediction Reduces Mismatch between Training and Testing Sets.Genes (Basel). 2024 Jul 23;15(8):969. doi: 10.3390/genes15080969. Genes (Basel). 2024. PMID: 39202329 Free PMC article.
-
In silico prediction of variant effects: promises and limitations for precision plant breeding.Theor Appl Genet. 2025 Jul 28;138(8):193. doi: 10.1007/s00122-025-04973-1. Theor Appl Genet. 2025. PMID: 40719915 Free PMC article. Review.
References
-
- Agrawal A. F., Whitlock M. C. (2012). Mutation load: The fitness of individuals in populations where deleterious alleles are abundant. Annu. Rev. Ecol. Evol. Syst. 43, 115–135. doi: 10.1146/annurev-ecolsys-110411-160257 - DOI
LinkOut - more resources
Full Text Sources