

iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria
- PMID: 37083735
- PMCID: PMC10155999
- DOI: 10.1371/journal.pbio.3002083
iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria
Abstract
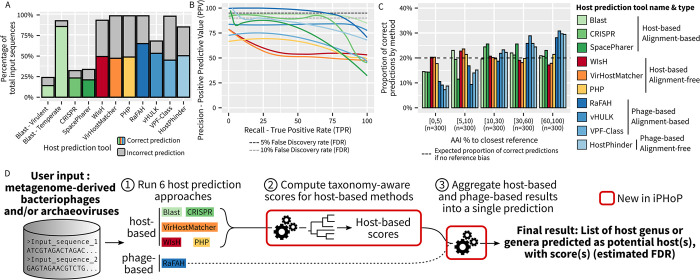
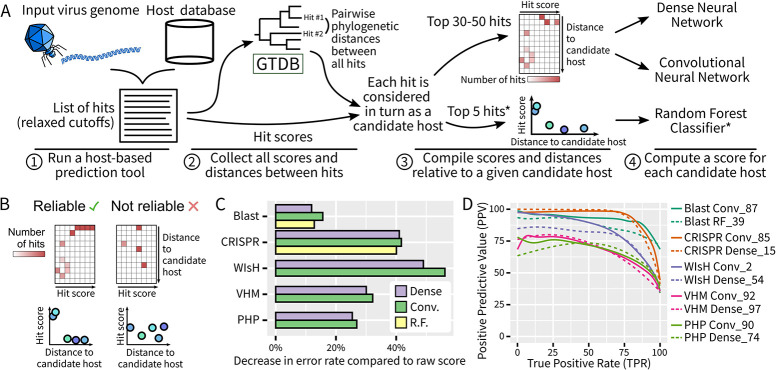
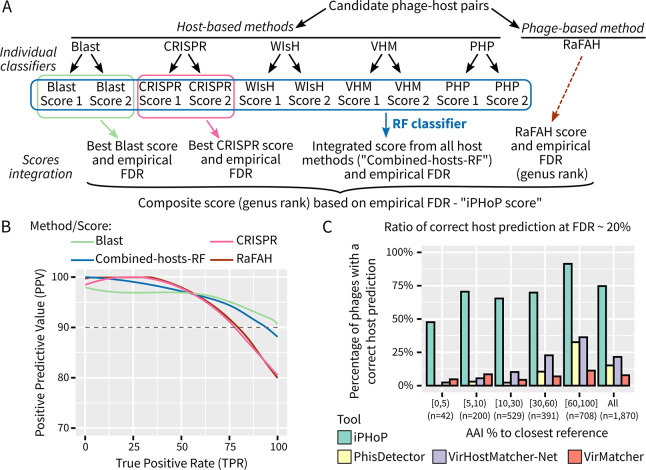
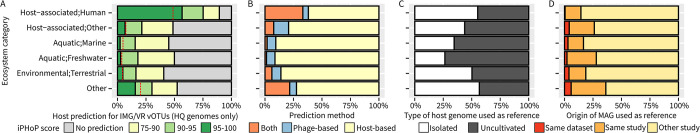
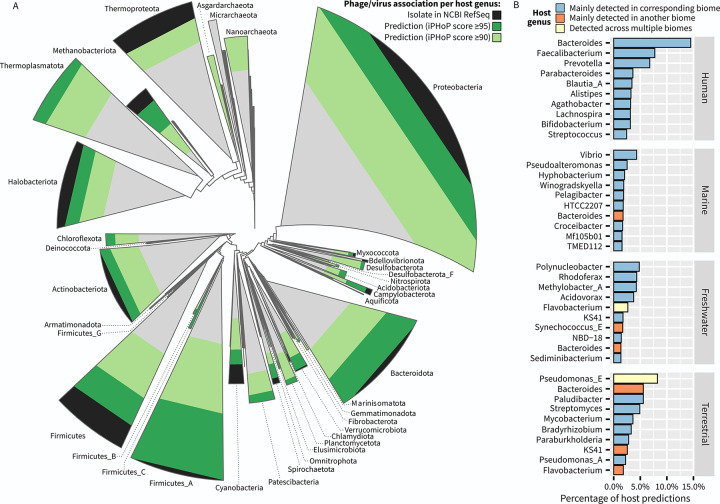
The extraordinary diversity of viruses infecting bacteria and archaea is now primarily studied through metagenomics. While metagenomes enable high-throughput exploration of the viral sequence space, metagenome-derived sequences lack key information compared to isolated viruses, in particular host association. Different computational approaches are available to predict the host(s) of uncultivated viruses based on their genome sequences, but thus far individual approaches are limited either in precision or in recall, i.e., for a number of viruses they yield erroneous predictions or no prediction at all. Here, we describe iPHoP, a two-step framework that integrates multiple methods to reliably predict host taxonomy at the genus rank for a broad range of viruses infecting bacteria and archaea, while retaining a low false discovery rate. Based on a large dataset of metagenome-derived virus genomes from the IMG/VR database, we illustrate how iPHoP can provide extensive host prediction and guide further characterization of uncultivated viruses.
Copyright: © 2023 Roux et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences.Microbiome. 2020 Jun 10;8(1):90. doi: 10.1186/s40168-020-00867-0. Microbiome. 2020. PMID: 32522236 Free PMC article.
-
A genomic catalog of Earth's microbiomes.Nat Biotechnol. 2021 Apr;39(4):499-509. doi: 10.1038/s41587-020-0718-6. Epub 2020 Nov 9. Nat Biotechnol. 2021. PMID: 33169036 Free PMC article.
-
Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks.Nat Biotechnol. 2019 Jun;37(6):632-639. doi: 10.1038/s41587-019-0100-8. Epub 2019 May 6. Nat Biotechnol. 2019. PMID: 31061483
-
An Introduction to Whole-Metagenome Shotgun Sequencing Studies.Methods Mol Biol. 2021;2243:107-122. doi: 10.1007/978-1-0716-1103-6_6. Methods Mol Biol. 2021. PMID: 33606255 Review.
-
Global overview and major challenges of host prediction methods for uncultivated phages.Curr Opin Virol. 2021 Aug;49:117-126. doi: 10.1016/j.coviro.2021.05.003. Epub 2021 Jun 12. Curr Opin Virol. 2021. PMID: 34126465 Review.
Cited by
-
Bacteriophages from treatment-naïve type 2 diabetes individuals drive an inflammatory response in human co-cultures of dendritic cells and T cells.Gut Microbes. 2024 Jan-Dec;16(1):2380747. doi: 10.1080/19490976.2024.2380747. Epub 2024 Jul 27. Gut Microbes. 2024. PMID: 39068518 Free PMC article.
-
Dispersal, habitat filtering, and eco-evolutionary dynamics as drivers of local and global wetland viral biogeography.ISME J. 2023 Nov;17(11):2079-2089. doi: 10.1038/s41396-023-01516-8. Epub 2023 Sep 21. ISME J. 2023. PMID: 37735616 Free PMC article.
-
zol and fai: large-scale targeted detection and evolutionary investigation of gene clusters.Nucleic Acids Res. 2025 Jan 24;53(3):gkaf045. doi: 10.1093/nar/gkaf045. Nucleic Acids Res. 2025. PMID: 39907107 Free PMC article.
-
Isolation and characterization of a roseophage representing a novel genus in the N4-like Rhodovirinae subfamily distributed in estuarine waters.BMC Genomics. 2025 Mar 25;26(1):295. doi: 10.1186/s12864-025-11463-7. BMC Genomics. 2025. PMID: 40133813 Free PMC article.
-
Engrafting gut bacteriophages have potential to modulate microbial metabolism in fecal microbiota transplantation.Microbiome. 2025 Jun 20;13(1):149. doi: 10.1186/s40168-025-02046-5. Microbiome. 2025. PMID: 40542451 Free PMC article.
