

Starch treatment improves the salivary proteome for subject identification purposes
- PMID: 37084127
- PMCID: PMC10944386
- DOI: 10.1007/s12024-023-00629-y
Starch treatment improves the salivary proteome for subject identification purposes
Abstract
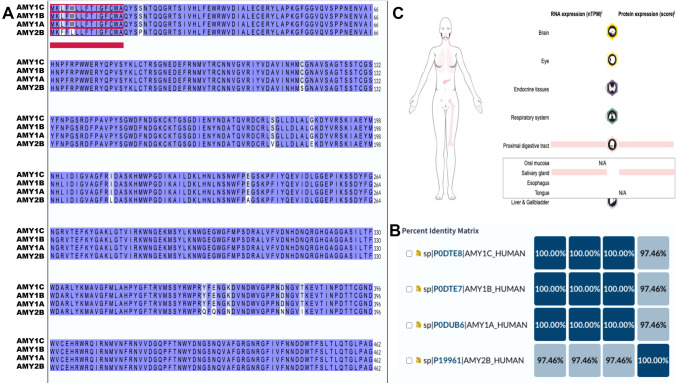
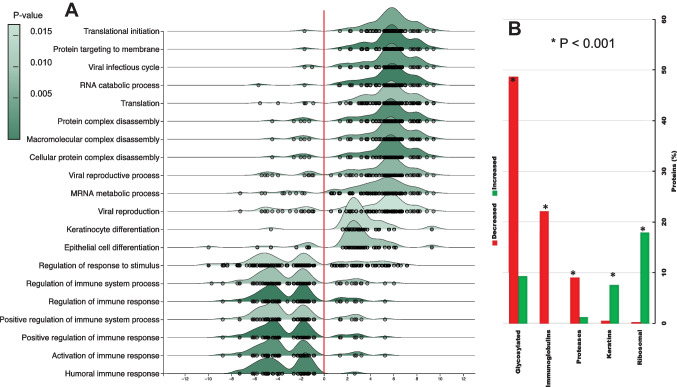
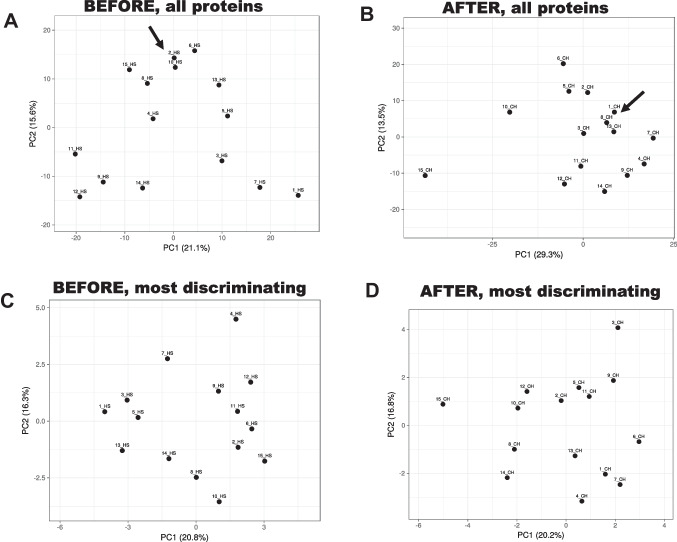
Identification of subjects, including perpetrators, is one of the most crucial goals of forensic science. Saliva is among the most common biological fluids found at crime scenes, containing identifiable components. DNA has been the most prominent identifier to date, but its analysis can be complex due to low DNA yields and issues preserving its integrity at the crime scene. Proteins are emerging as viable candidates for subject identification. Previous work has shown that the salivary proteome of the least-abundant proteins may be helpful for subject identification, but more optimized techniques are needed. Among them is removing the most abundant proteins, such as salivary α-amylase. Starch treatment of saliva samples elicited the removal of this enzyme and that of glycosylated, low-molecular-weight proteins, proteases, and immunoglobulins, resulting in a saliva proteome profile enriched with a subset of proteins, allowing a more reliable and nuanced subject identification.
Keywords: Forensic identification; Mass spectrometry; Method; Proteome; Saliva; Starch.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Anzai-Kanto E, Hirata MH, Hirata RD, Nunes FD, Melani RF, Oliveira RN. DNA extraction from human saliva deposited on skin and its use in forensic identification procedures. Braz Oral Res. 2005;19(3):216–22. Available from: https://www.ncbi.nlm.nih.gov/pubmed/16308611. - PubMed
-
- Chatterjee S. Saliva as a forensic tool. J Forensic Dent Sci. 2019;11(1):1–4. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31680748. - PMC - PubMed
-
- Van Steendam K, De Ceuleneer M, Dhaenens M, Van Hoofstat D, Deforce D. Mass spectrometry-based proteomics as a tool to identify biological matrices in forensic science. Int J Legal Med. 2013;127(2):287–98. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22843116. - PMC - PubMed
-
- Toennes SW, Steinmeyer S, Maurer HJ, Moeller MR, Kauert GF. Screening for drugs of abuse in oral fluid--correlation of analysis results with serum in forensic cases. J Anal Toxicol. 2005;29(1):22–7. Available from: https://www.ncbi.nlm.nih.gov/pubmed/15808009. - PubMed
-
- Grassl N, Kulak NA, Pichler G, Geyer PE, Jung J, Schubert S, Sinitcyn P, Cox J, Mann M. Ultra-deep and quantitative saliva proteome reveals dynamics of the oral microbiome. Genome Med. 2016;8(1):44. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27102203. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
