

Spectrum of clonal hematopoiesis in VEXAS syndrome
- PMID: 37084382
- PMCID: PMC10375269
- DOI: 10.1182/blood.2022018774
Spectrum of clonal hematopoiesis in VEXAS syndrome
Abstract
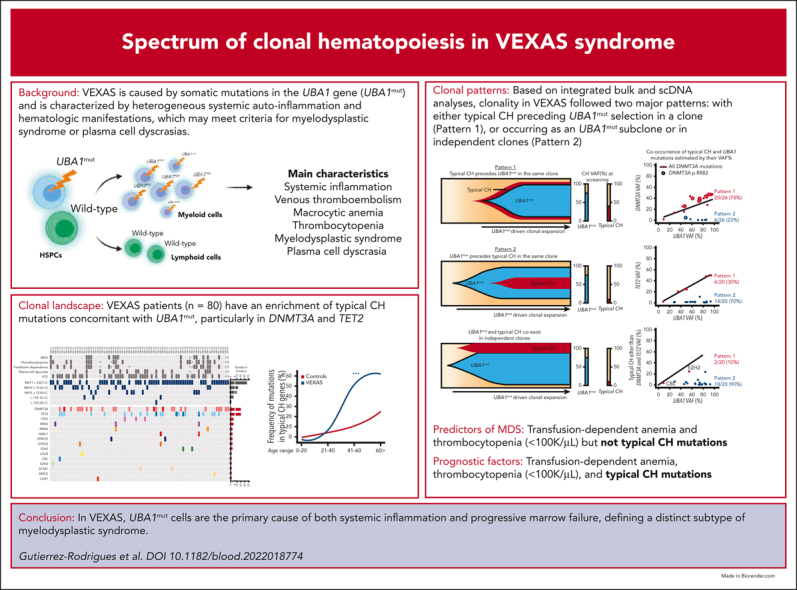
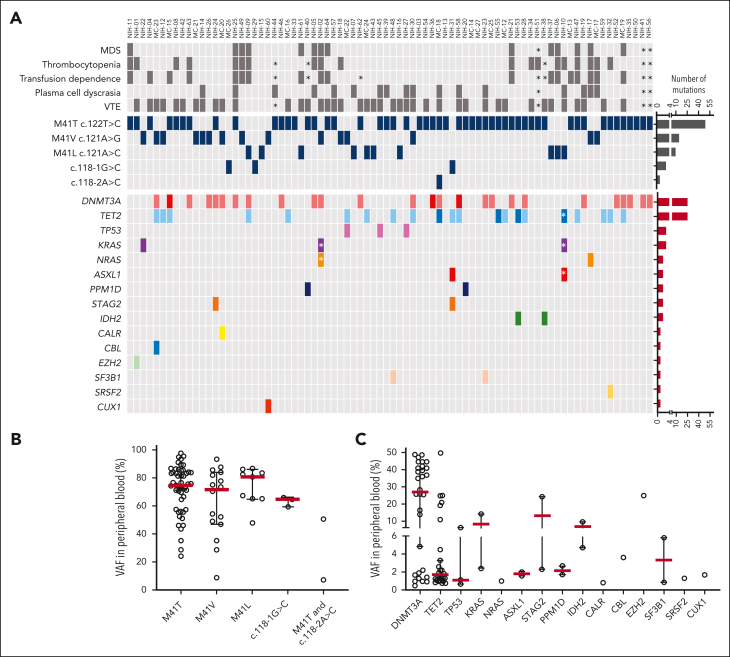
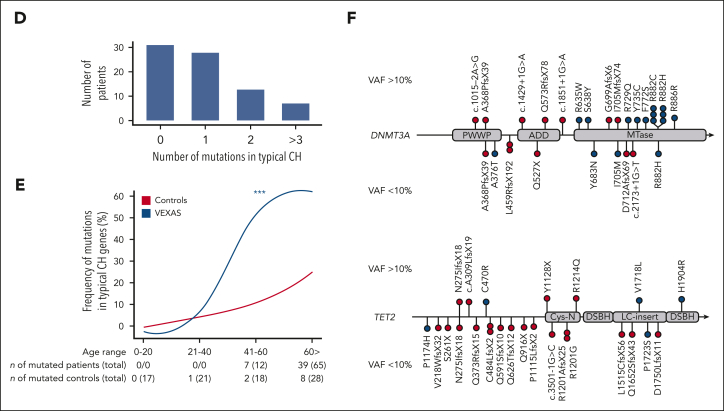
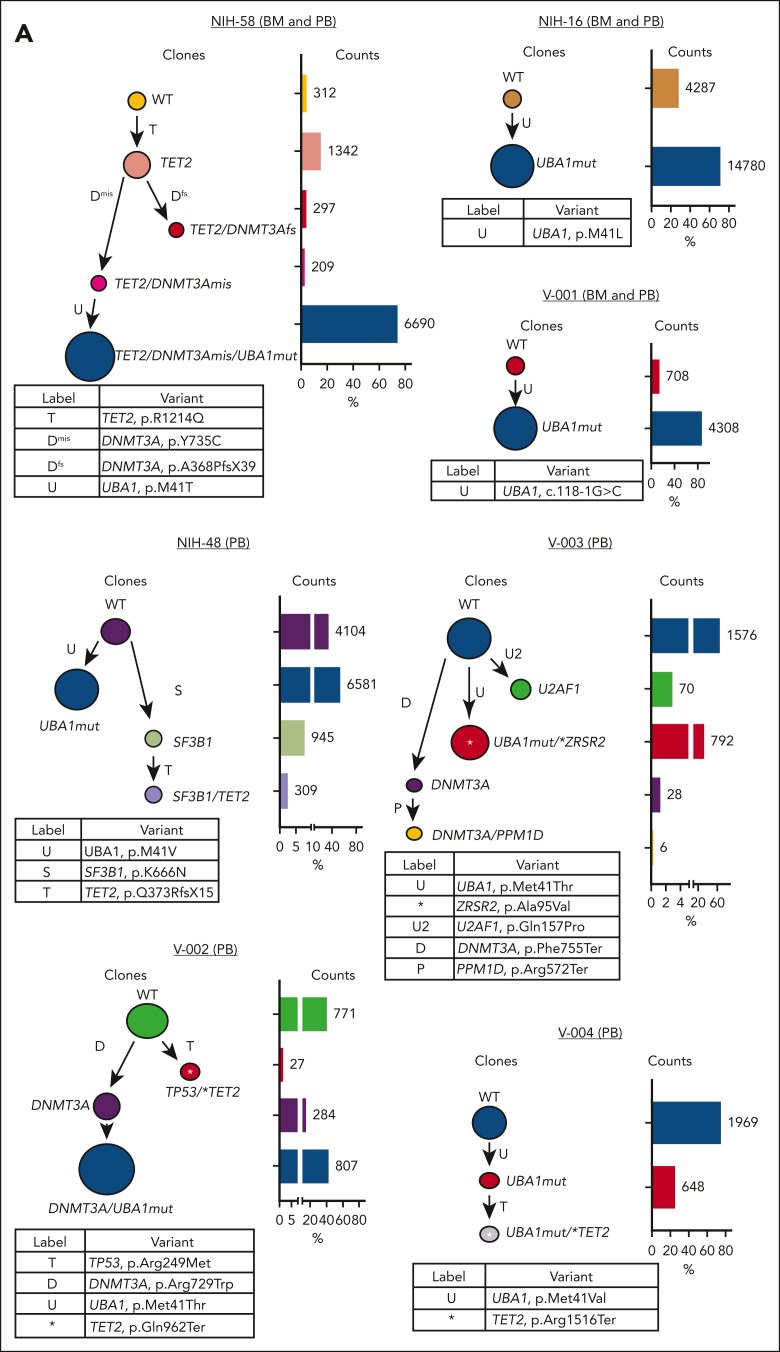
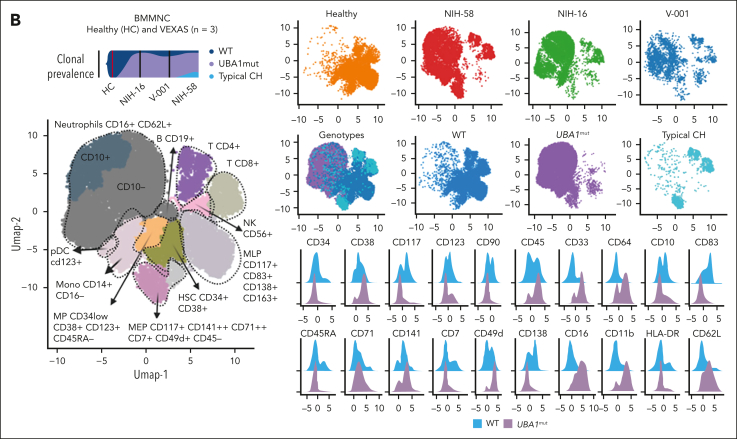
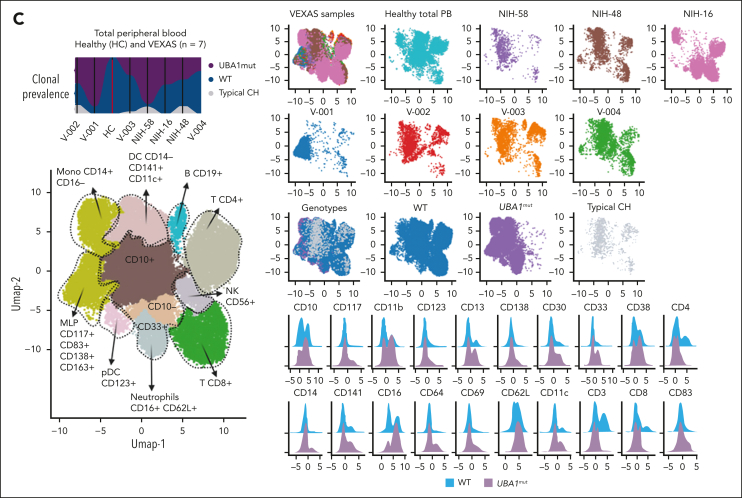
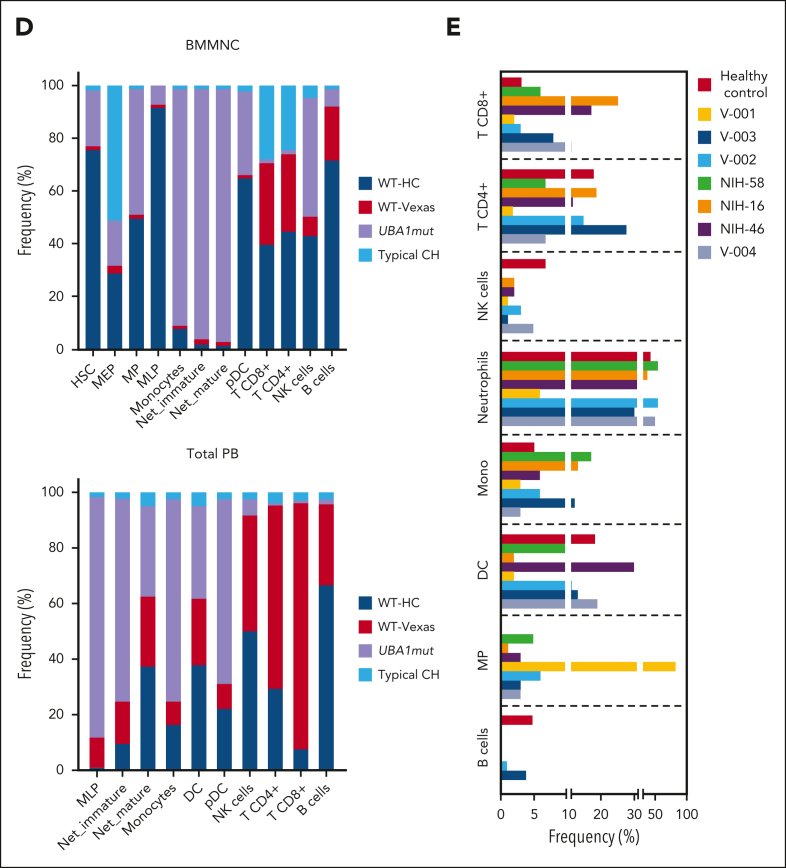
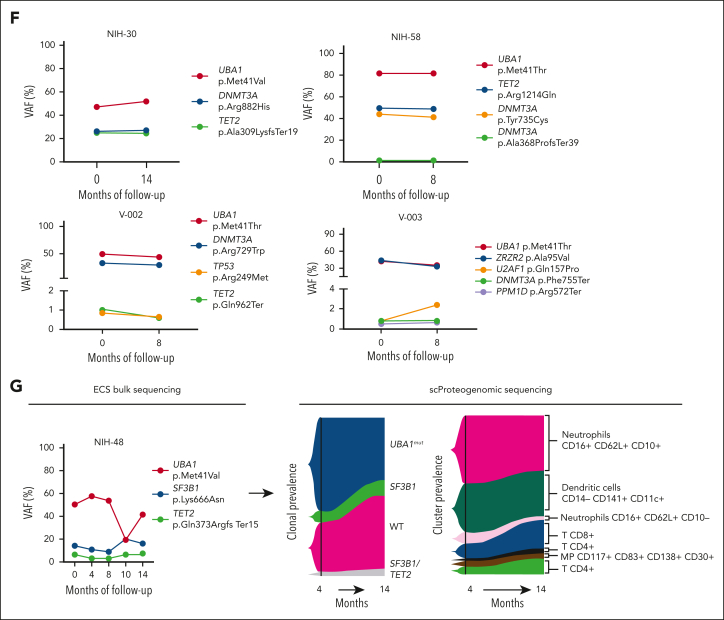
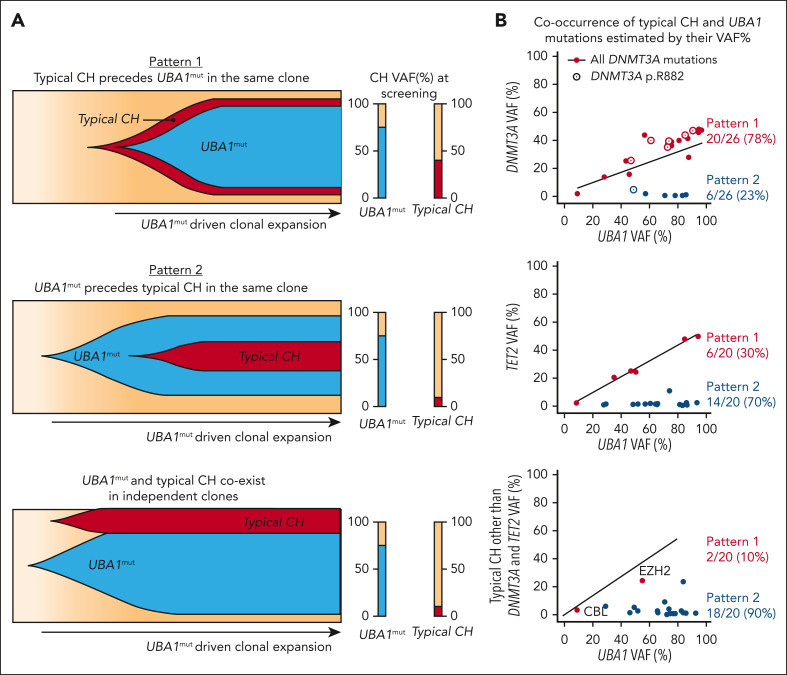
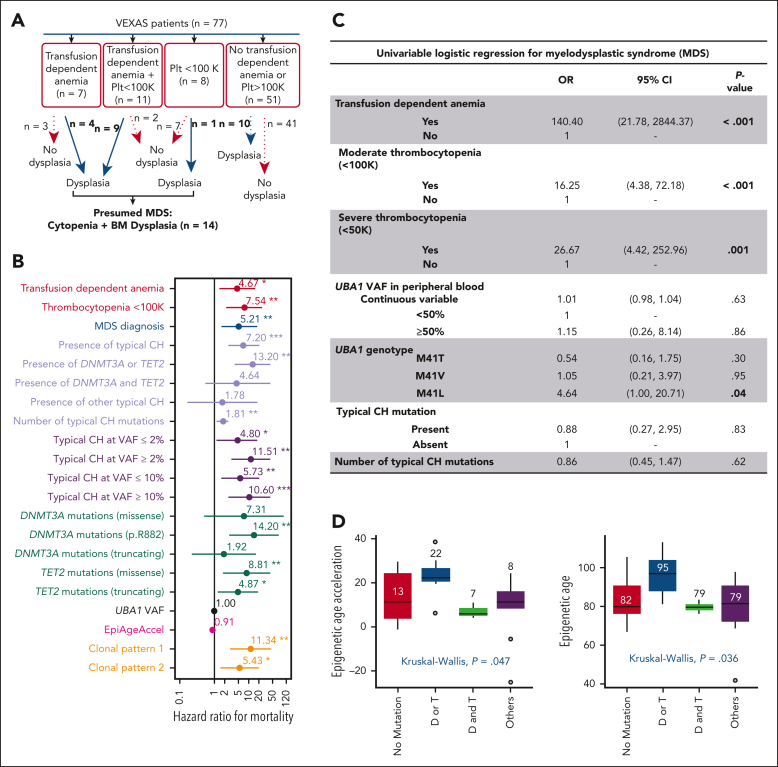
Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome is caused by somatic mutations in UBA1 (UBA1mut) and characterized by heterogenous systemic autoinflammation and progressive hematologic manifestations, meeting criteria for myelodysplastic syndrome (MDS) and plasma cell dyscrasias. The landscape of myeloid-related gene mutations leading to typical clonal hematopoiesis (CH) in these patients is unknown. Retrospectively, we screened 80 patients with VEXAS for CH in their peripheral blood (PB) and correlated the findings with clinical outcomes in 77 of them. UBA1mut were most common at hot spot p.M41 (median variant allele frequency [VAF] = 75%). Typical CH mutations cooccurred with UBA1mut in 60% of patients, mostly in DNMT3A and TET2, and were not associated with inflammatory or hematologic manifestations. In prospective single-cell proteogenomic sequencing (scDNA), UBA1mut was the dominant clone, present mostly in branched clonal trajectories. Based on integrated bulk and scDNA analyses, clonality in VEXAS followed 2 major patterns: with either typical CH preceding UBA1mut selection in a clone (pattern 1) or occurring as an UBA1mut subclone or in independent clones (pattern 2). VAF in the PB differed markedly between DNMT3A and TET2 clones (median VAF of 25% vs 1%). DNMT3A and TET2 clones associated with hierarchies representing patterns 1 and 2, respectively. Overall survival for all patients was 60% at 10 years. Transfusion-dependent anemia, moderate thrombocytopenia, and typical CH mutations, each correlated with poor outcome. In VEXAS, UBA1mut cells are the primary cause of systemic inflammation and marrow failure, being a new molecularly defined somatic entity associated with MDS. VEXAS-associated MDS is distinct from classical MDS in its presentation and clinical course.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing interests.
Figures
Comment in
-
VEXAS: walking on the edge of malignancy.Blood. 2023 Jul 20;142(3):214-215. doi: 10.1182/blood.2023020772. Blood. 2023. PMID: 37471107 No abstract available.
References
-
- Georgin-Lavialle S, Terrier B, Guedon AF, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. 2022;186(3):564–574. - PubMed
-
- Bourbon E, Heiblig M, Gerfaud Valentin M, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. 2021;137(26):3682–3684. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
