

Proteomic discovery of chemical probes that perturb protein complexes in human cells
- PMID: 37084731
- PMCID: PMC10198961
- DOI: 10.1016/j.molcel.2023.03.026
Proteomic discovery of chemical probes that perturb protein complexes in human cells
Abstract
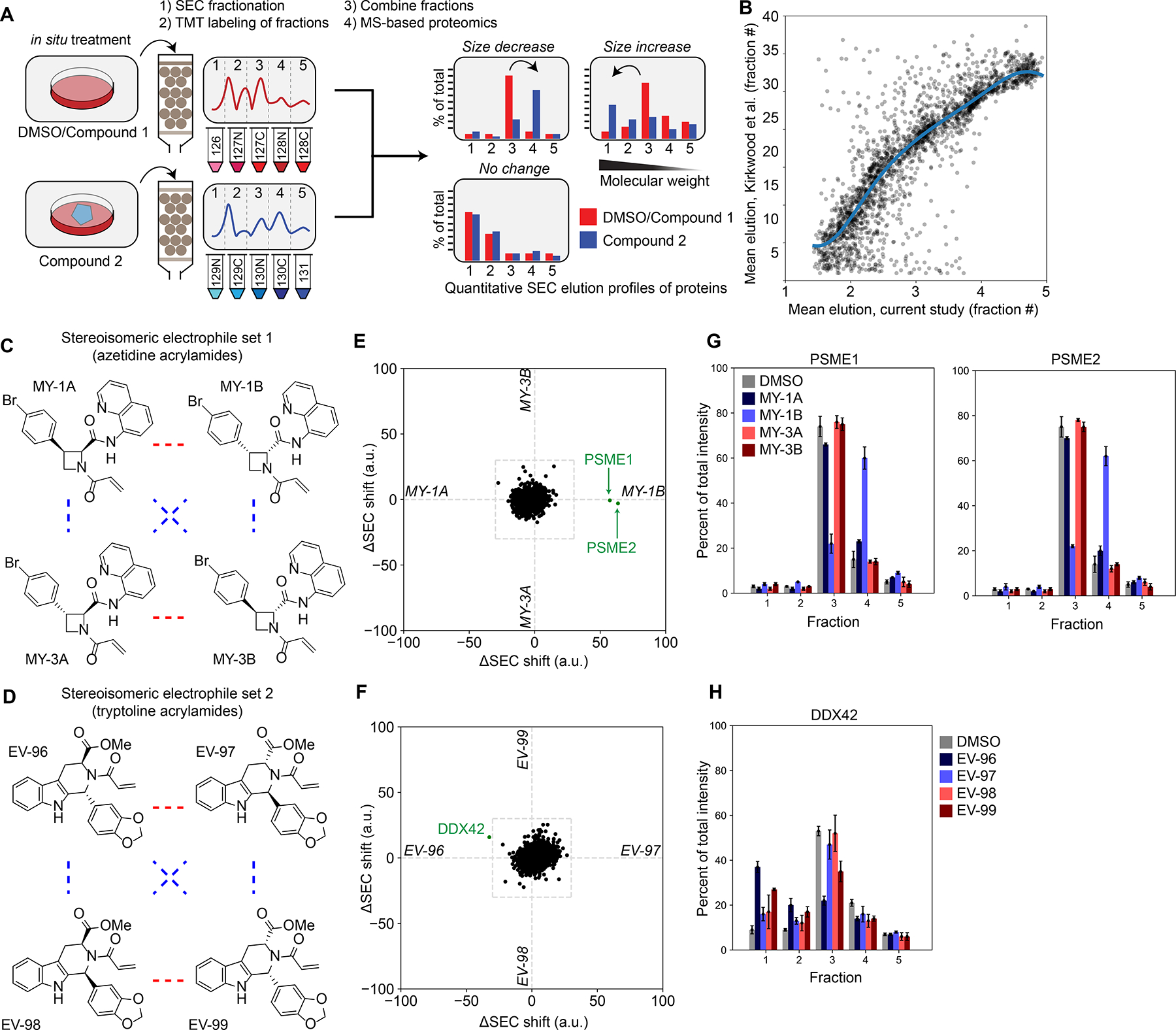
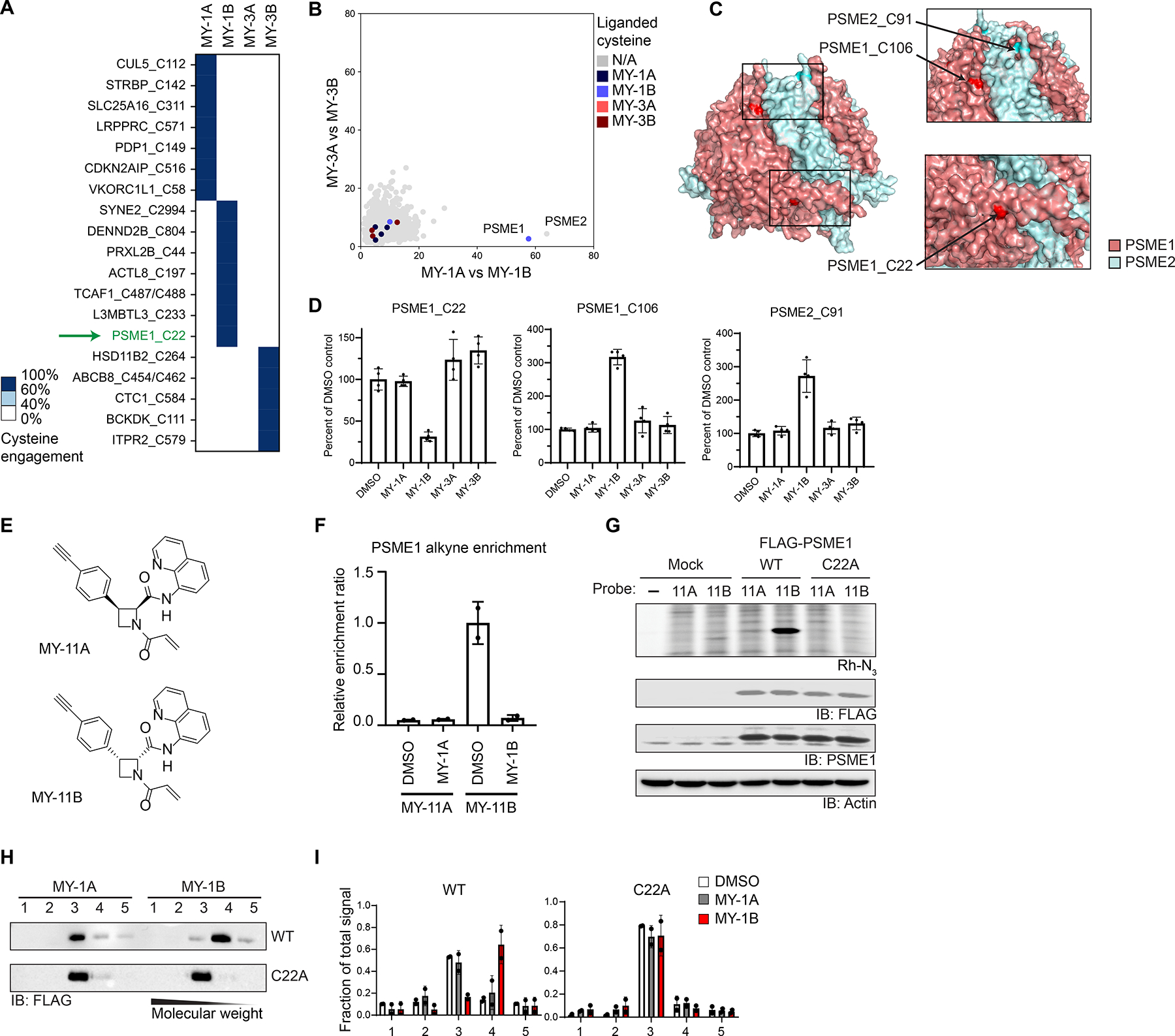
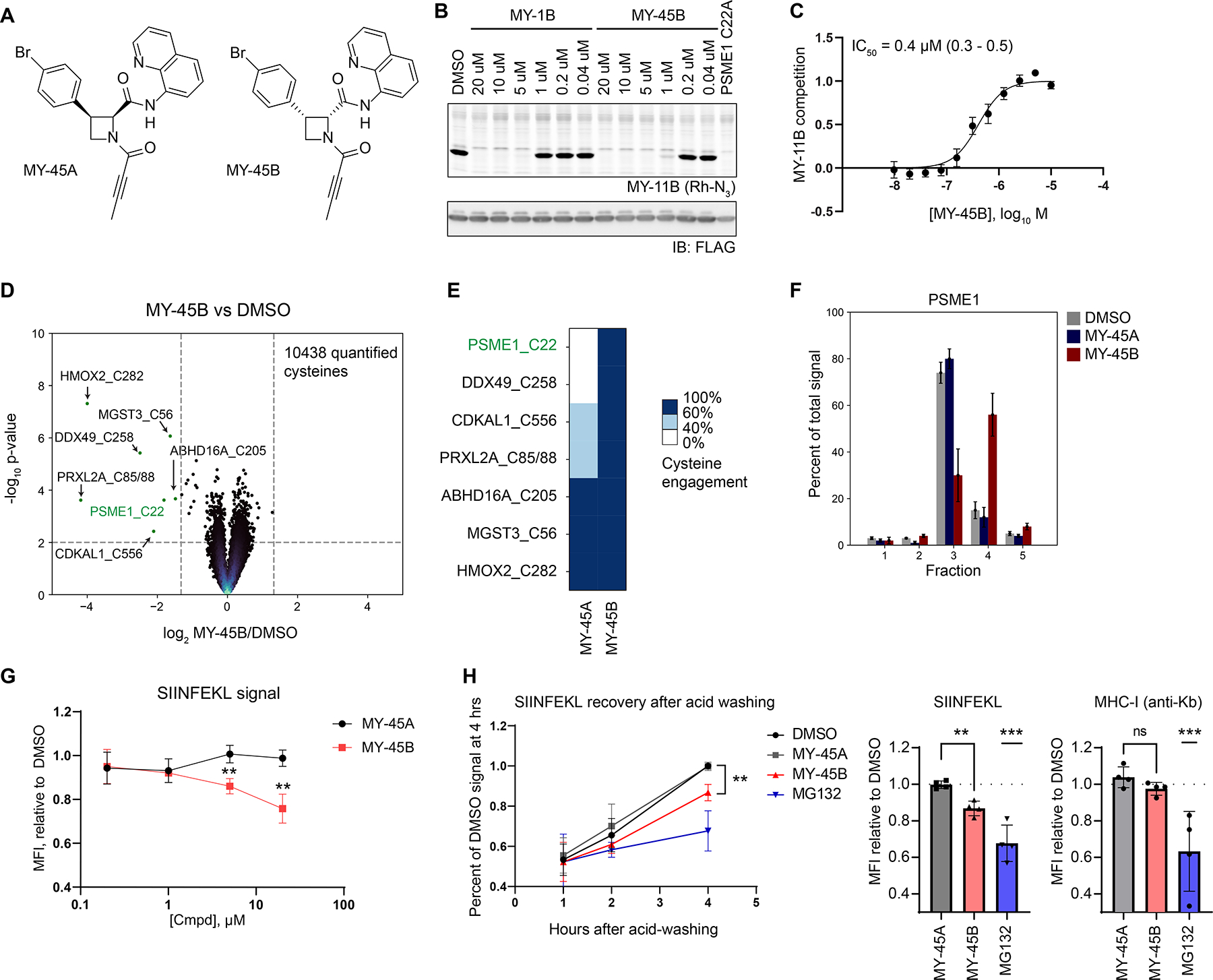
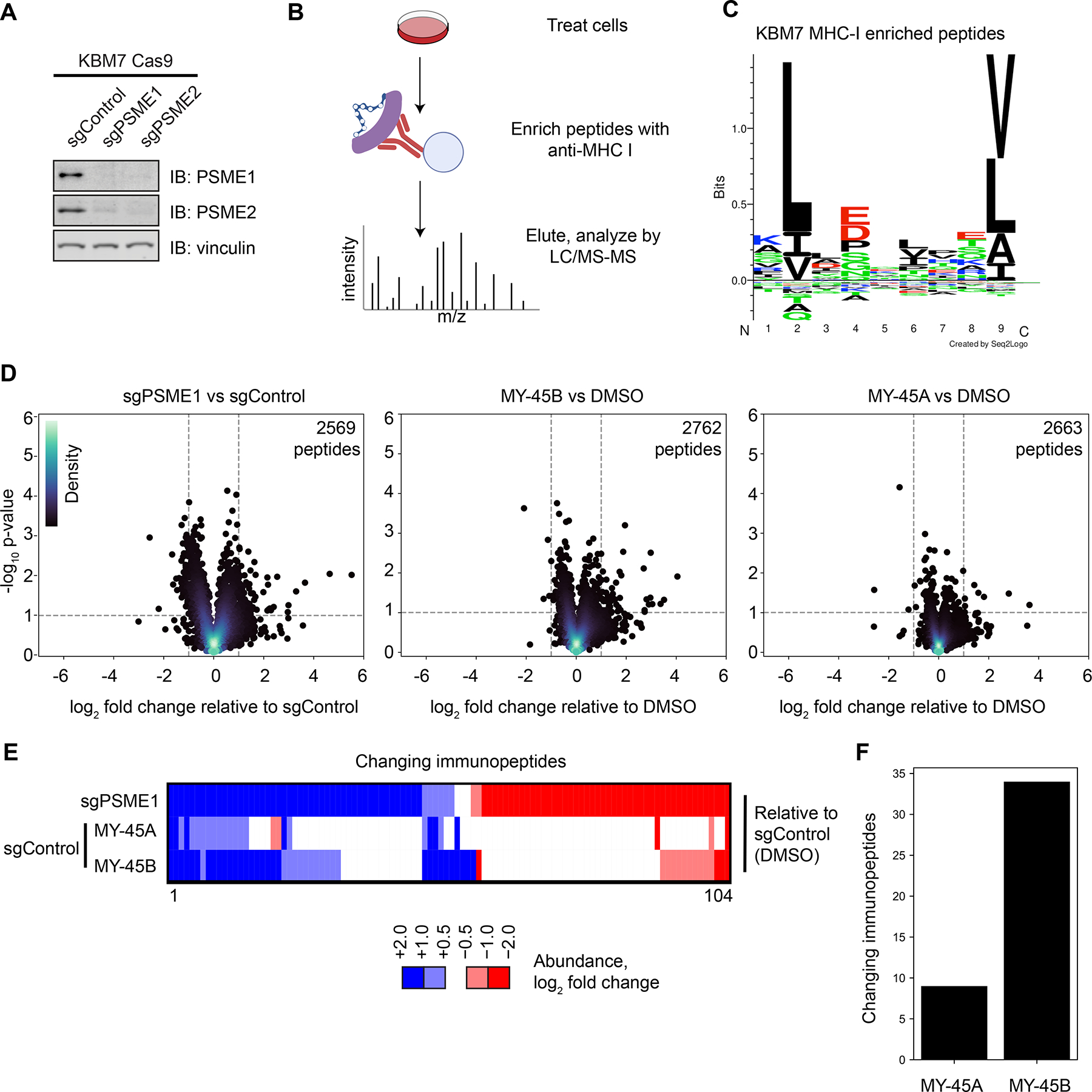
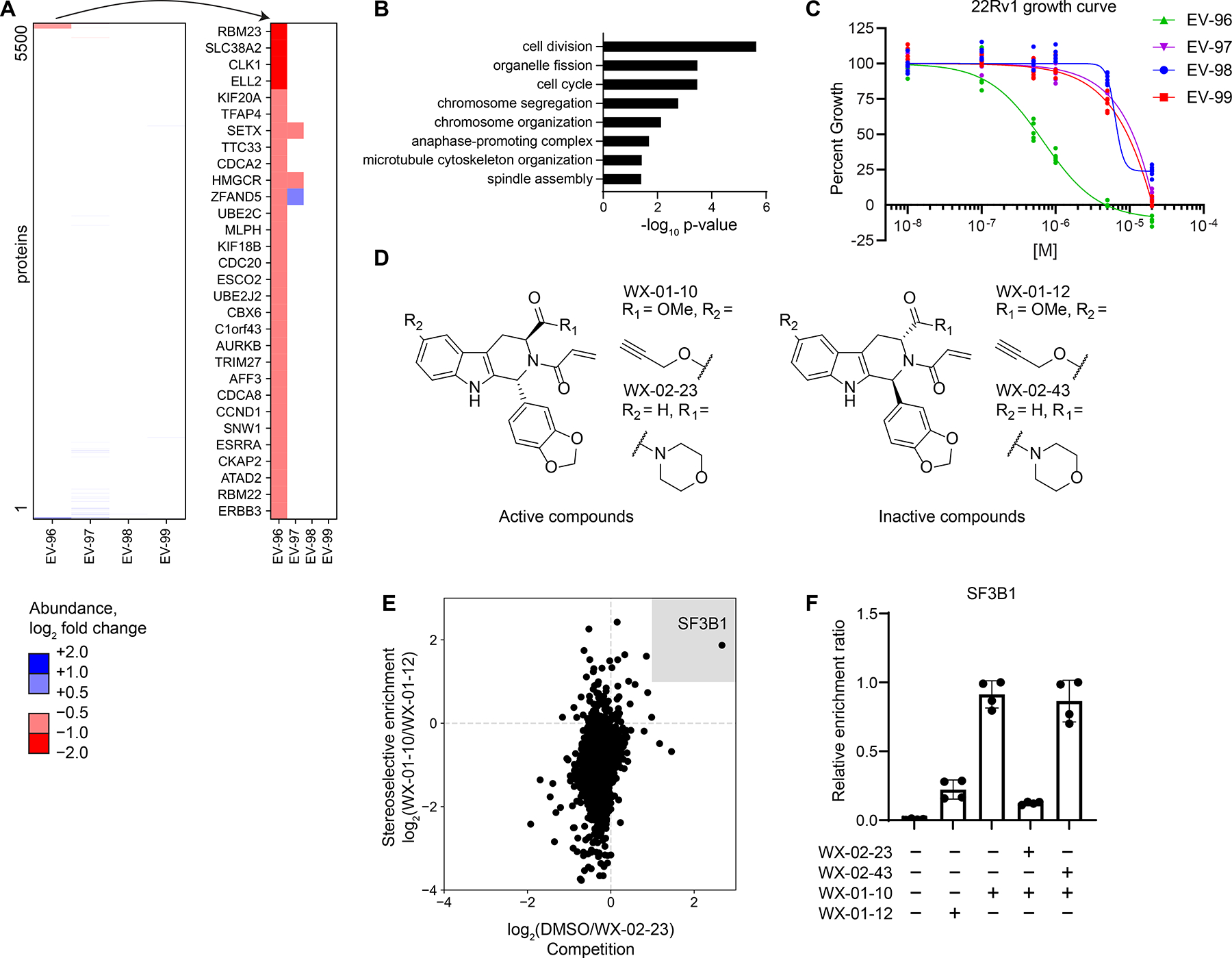
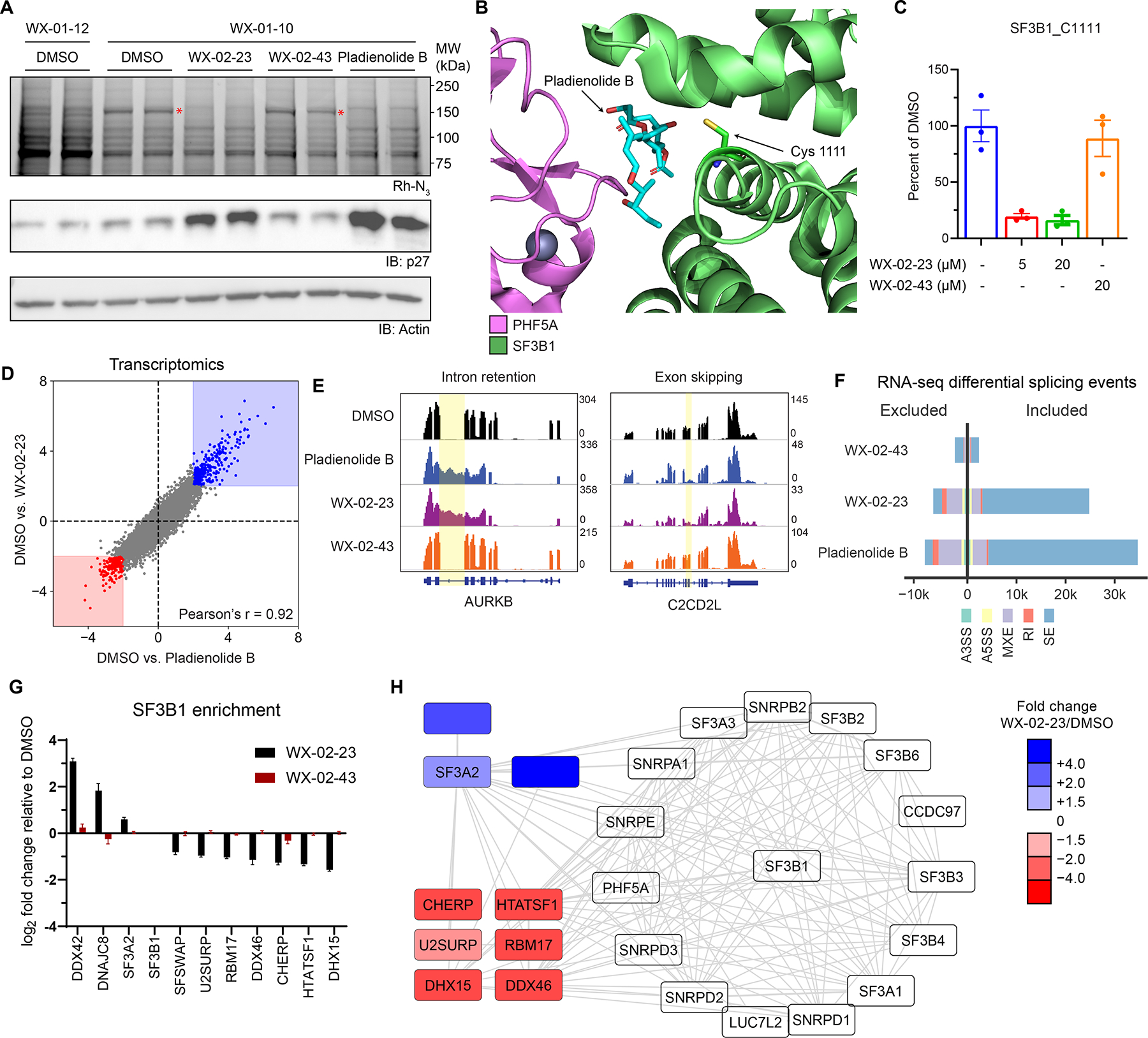
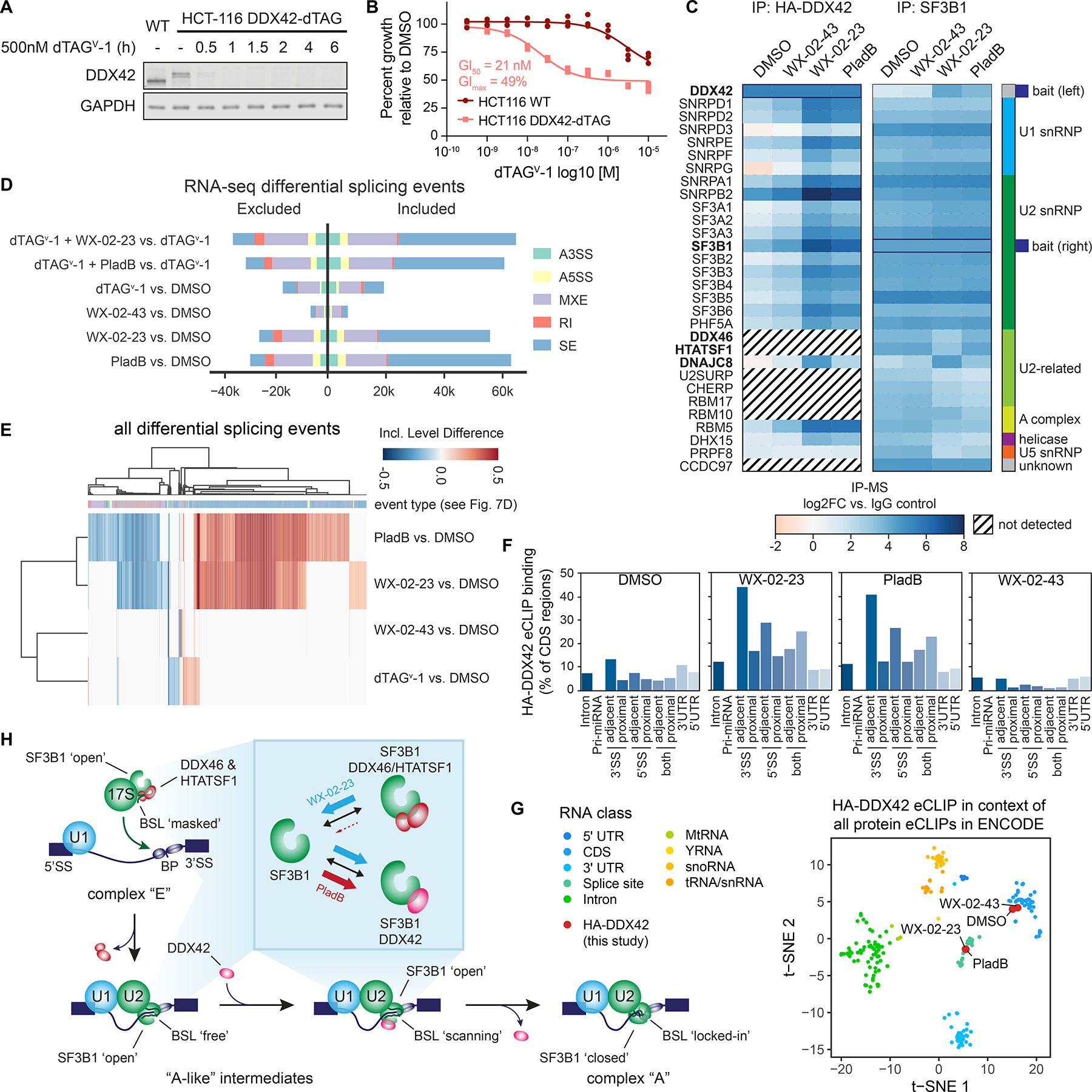
Most human proteins lack chemical probes, and several large-scale and generalizable small-molecule binding assays have been introduced to address this problem. How compounds discovered in such "binding-first" assays affect protein function, nonetheless, often remains unclear. Here, we describe a "function-first" proteomic strategy that uses size exclusion chromatography (SEC) to assess the global impact of electrophilic compounds on protein complexes in human cells. Integrating the SEC data with cysteine-directed activity-based protein profiling identifies changes in protein-protein interactions that are caused by site-specific liganding events, including the stereoselective engagement of cysteines in PSME1 and SF3B1 that disrupt the PA28 proteasome regulatory complex and stabilize a dynamic state of the spliceosome, respectively. Our findings thus show how multidimensional proteomic analysis of focused libraries of electrophilic compounds can expedite the discovery of chemical probes with site-specific functional effects on protein complexes in human cells.
Keywords: activity-based protein profiling; chemical probe; covalent; cysteine; proteasome; protein complexes; proteomics; size-exclusion chromatography; spliceosome.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests G.M.S., V.F.V., and L.R.W. are employees of Vividion Therapeutics, and B.F.C. is a founder and member of the Board of Directors of Vividion Therapeutics. G.W.Y. is a co-founder, member of the Board of Directors, on the SAB, equity holder, and paid consultant for Locanabio and Eclipse BioInnovations. G.W.Y.’s interests have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. A US provisional patent has been filed related to the work disclosed in this manuscript.
Figures
Comment in
-
Discovery of chemical probes that perturb protein complexes using size exclusion chromatography and chemical proteomics.Mol Cell. 2023 May 18;83(10):1544-1546. doi: 10.1016/j.molcel.2023.04.028. Mol Cell. 2023. PMID: 37207621
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
