

Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers
- PMID: 37085492
- PMCID: PMC10121712
- DOI: 10.1038/s41467-023-37437-5
Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers
Abstract
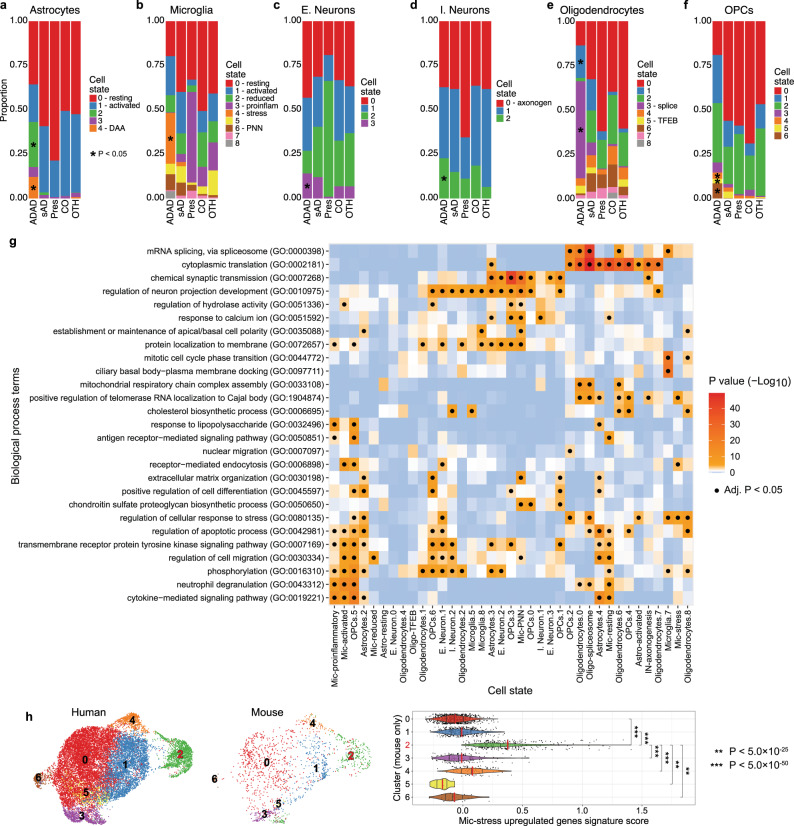
Genetic studies of Alzheimer disease (AD) have prioritized variants in genes related to the amyloid cascade, lipid metabolism, and neuroimmune modulation. However, the cell-specific effect of variants in these genes is not fully understood. Here, we perform single-nucleus RNA-sequencing (snRNA-seq) on nearly 300,000 nuclei from the parietal cortex of AD autosomal dominant (APP and PSEN1) and risk-modifying variant (APOE, TREM2 and MS4A) carriers. Within individual cell types, we capture genes commonly dysregulated across variant groups. However, specific transcriptional states are more prevalent within variant carriers. TREM2 oligodendrocytes show a dysregulated autophagy-lysosomal pathway, MS4A microglia have dysregulated complement cascade genes, and APOEε4 inhibitory neurons display signs of ferroptosis. All cell types have enriched states in autosomal dominant carriers. We leverage differential expression and single-nucleus ATAC-seq to map GWAS signals to effector cell types including the NCK2 signal to neurons in addition to the initially proposed microglia. Overall, our results provide insights into the transcriptional diversity resulting from AD genetic architecture and cellular heterogeneity. The data can be explored on the online browser ( http://web.hararilab.org/SNARE/ ).
© 2023. The Author(s).
Conflict of interest statement
J.C.M. is a consultant for the Barcelona Brain Research Center (BBRC) and the TS Srinivasan Advisory Board. J.C.M. is an advisory board member for the Cure Alzheimer’s Fund Research Strategy Council. R.J.B. maintains an equity ownership interest and is a member of the advisory board of C2N Diagnostics. Unrelated to this article, R.J.B. serves as the principal investigator of the DIAN-TU, which the Alzheimer’s Association supports, GHR Foundation, an anonymous organization, and the DIAN-TU Pharma Consortium (Active: Eli Lilly and Company/Avid Radiopharmaceuticals, F. Hoffman-La Roche/Genentech, Biogen, Eisai, and Janssen. Previous: Abbvie, Amgen, AstraZeneca, Forum, Mithridion, Novartis, Pfizer, Sanofi, and United Neuroscience). In addition, in-kind support has been received from CogState and Signant Health. Unrelated to this article, R.J.B. has submitted the US nonprovisional patent application “Methods for Measuring the Metabolism of CNS Derived Biomolecules in Vivo” and provisional patent application “Plasma Based Methods for Detecting CNS Amyloid Deposition.” E.M. receives research support from the NIA, Hoffman-La Roche, and Eli Lilly, is a member of advisory boards for Eli Lilly, Alector, and the NIA, and holds a leadership role in Fondation Alzheimer and Alzamend. C.X. is a consultant for DIADEM and a member of the advisory board for the University of Wisconsin ADRC. A.G. receives royalties from Athena Diagnostics and Taconic Biosciences, is a consultant for Genentech SAB and AbbVie and holds stock or stock options in Cognition Therapeutics and Denali Therapeutics. M.F. receives research support from Eli Lilly and Company, Hoffmann-La Roche, Avanir, Biogen, Cognition Therapies, Green Valley, Otsuka, Neurotrope Biosciences, AZTherapies, Athira, Ionis, and Lexeo, and is a member of advisory boards for Oligomerik and T3D. The remaining authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
- U01 AG046152/AG/NIA NIH HHS/United States
- R01 AG062734/AG/NIA NIH HHS/United States
- R01 AG030146/AG/NIA NIH HHS/United States
- U01 AG072464/AG/NIA NIH HHS/United States
- T32 AG058518/AG/NIA NIH HHS/United States
- R01 AG074012/AG/NIA NIH HHS/United States
- RC2 AG036547/AG/NIA NIH HHS/United States
- R01 NS118146/NS/NINDS NIH HHS/United States
- U01 AG046161/AG/NIA NIH HHS/United States
- R01 AG048015/AG/NIA NIH HHS/United States
- R01 AG057914/AG/NIA NIH HHS/United States
- R56 AG067764/AG/NIA NIH HHS/United States
- P30 AG066444/AG/NIA NIH HHS/United States
- R01 AG057777/AG/NIA NIH HHS/United States
- RF1 AG071706/AG/NIA NIH HHS/United States
- U01 AG058922/AG/NIA NIH HHS/United States
- R01 AG044546/AG/NIA NIH HHS/United States
- U24 AG021886/AG/NIA NIH HHS/United States
- U01 AG061356/AG/NIA NIH HHS/United States
- U01 AG032984/AG/NIA NIH HHS/United States
- R01 AG078964/AG/NIA NIH HHS/United States
- P01 AG003991/AG/NIA NIH HHS/United States
- P01 AG026276/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- RF1 AG058501/AG/NIA NIH HHS/United States
- R01 AG036042/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- R21 NS127211/NS/NINDS NIH HHS/United States
- RF1 AG057473/AG/NIA NIH HHS/United States
- RF1 AG044546/AG/NIA NIH HHS/United States
- U19 AG032438/AG/NIA NIH HHS/United States
- R01 AG036836/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
