

A systematic outbreak investigation of SARS-CoV-2 transmission clusters in a tertiary academic care center
- PMID: 37085891
- PMCID: PMC10119817
- DOI: 10.1186/s13756-023-01242-y
A systematic outbreak investigation of SARS-CoV-2 transmission clusters in a tertiary academic care center
Abstract
Background: We sought to decipher transmission pathways in healthcare-associated infections with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) within our hospital by epidemiological work-up and complementary whole genome sequencing (WGS). We report the findings of the four largest epidemiologic clusters of SARS-CoV-2 transmission occurring during the second wave of the pandemic from 11/2020 to 12/2020.
Methods: At the University Hospital Basel, Switzerland, systematic outbreak investigation is initiated at detection of any nosocomial case of SARS-CoV-2 infection, as confirmed by polymerase chain reaction, occurring more than five days after admission. Clusters of nosocomial infections, defined as the detection of at least two positive patients and/or healthcare workers (HCWs) within one week with an epidemiological link, were further investigated by WGS on respective strains.
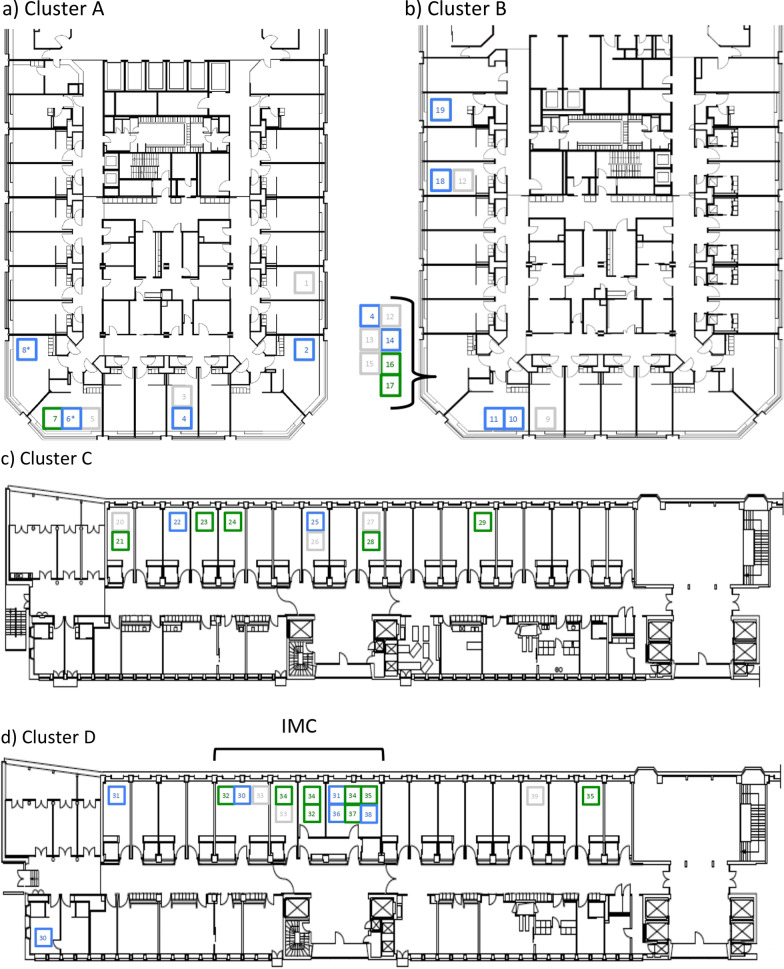
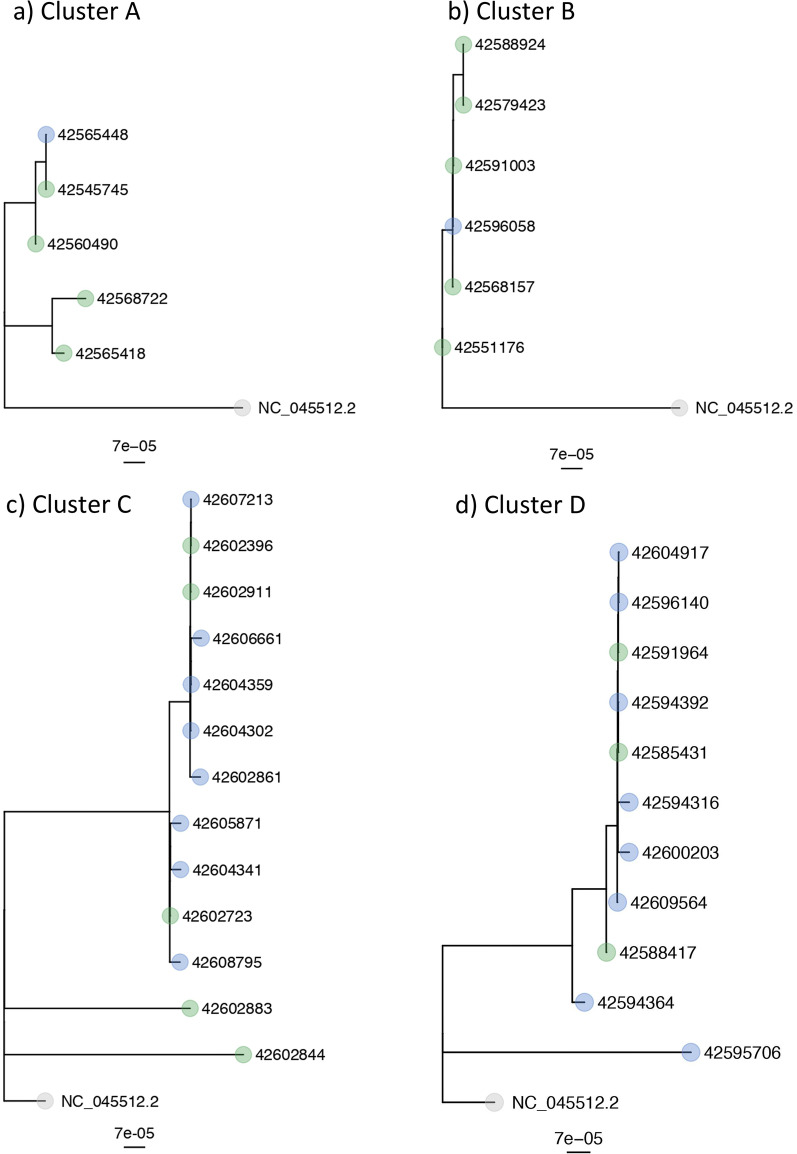
Results: The four epidemiologic clusters included 40 patients and 60 HCWs. Sequencing data was available for 70% of all involved cases (28 patients and 42 HCWs), confirmed epidemiologically suspected in house transmission in 33 cases (47.1% of sequenced cases) and excluded transmission in the remaining 37 cases (52.9%). Among cases with identical strains, epidemiologic work-up suggested transmission mainly through a ward-based exposure (24/33, 72.7%), more commonly affecting HCWs (16/24, 66.7%) than patients (8/24, 33.3%), followed by transmission between patients (6/33, 18.2%), and among HCWs and patients (3/33, 9.1%, respectively two HCWs and one patient).
Conclusions: Phylogenetic analyses revealed important insights into transmission pathways supporting less than 50% of epidemiologically suspected SARS-CoV-2 transmissions. The remainder of cases most likely reflect community-acquired infection randomly detected by outbreak investigation. Notably, most transmissions occurred between HCWs, possibly indicating lower perception of the risk of infection during contacts among HCWs.
Keywords: COVID-19; Epidemiologic cluster; Nosocomial outbreaks; Outbreak investigation; SARS-CoV-2 cluster; Whole genome sequencing.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Combining epidemiological data and whole genome sequencing to understand SARS-CoV-2 transmission dynamics in a large tertiary care hospital during the first COVID-19 wave in The Netherlands focusing on healthcare workers.Antimicrob Resist Infect Control. 2023 May 10;12(1):46. doi: 10.1186/s13756-023-01247-7. Antimicrob Resist Infect Control. 2023. PMID: 37165456 Free PMC article.
-
Explosive nosocomial outbreak of SARS-CoV-2 in a rehabilitation clinic: the limits of genomics for outbreak reconstruction.J Hosp Infect. 2021 Nov;117:124-134. doi: 10.1016/j.jhin.2021.07.013. Epub 2021 Aug 27. J Hosp Infect. 2021. PMID: 34461177 Free PMC article.
-
Reconstruction of transmission chains of SARS-CoV-2 amidst multiple outbreaks in a geriatric acute-care hospital: a combined retrospective epidemiological and genomic study.Elife. 2022 Jul 19;11:e76854. doi: 10.7554/eLife.76854. Elife. 2022. PMID: 35850933 Free PMC article.
-
Nosocomial Outbreak of SARS-CoV-2 in a Hospital Ward during the Omicron Variant-Dominant Wave with a Review of the Relevant Literature.Jpn J Infect Dis. 2024 Sep 19;77(5):253-259. doi: 10.7883/yoken.JJID.2023.464. Epub 2024 May 31. Jpn J Infect Dis. 2024. PMID: 38825458 Review.
-
Nosocomial transmission and outbreaks of coronavirus disease 2019: the need to protect both patients and healthcare workers.Antimicrob Resist Infect Control. 2021 Jan 6;10(1):7. doi: 10.1186/s13756-020-00875-7. Antimicrob Resist Infect Control. 2021. PMID: 33407833 Free PMC article. Review.
Cited by
-
AUTO-TUNE: SELECTING THE DISTANCE THRESHOLD FOR INFERRING HIV TRANSMISSION CLUSTERS.bioRxiv [Preprint]. 2024 Mar 14:2024.03.11.584522. doi: 10.1101/2024.03.11.584522. bioRxiv. 2024. Update in: Front Bioinform. 2024 Jul 10;4:1400003. doi: 10.3389/fbinf.2024.1400003. PMID: 38559140 Free PMC article. Updated. Preprint.
-
AUTO-TUNE: selecting the distance threshold for inferring HIV transmission clusters.Front Bioinform. 2024 Jul 10;4:1400003. doi: 10.3389/fbinf.2024.1400003. eCollection 2024. Front Bioinform. 2024. PMID: 39086842 Free PMC article.
-
Secondary attack rate following on-site isolation of patients with suspected COVID-19 in multiple-bed rooms.Antimicrob Resist Infect Control. 2024 Jul 6;13(1):73. doi: 10.1186/s13756-024-01430-4. Antimicrob Resist Infect Control. 2024. PMID: 38971822 Free PMC article.
-
Risk of SARS-CoV-2 infection before and after the Omicron wave in a cohort of healthcare workers in Ontario, Canada.BMC Infect Dis. 2025 Feb 7;25(1):183. doi: 10.1186/s12879-025-10580-8. BMC Infect Dis. 2025. PMID: 39920611 Free PMC article.
-
Utilizing Whole Genome Sequencing to Investigate a COVID-19 Cluster Among Healthcare Workers in a Tertiary Care Facility in the Philippines: Insights and Implications for Infection Prevention and Control.Clin Infect Dis. 2025 Jul 18;80(6):1262-1268. doi: 10.1093/cid/ciaf057. Clin Infect Dis. 2025. PMID: 40590532 Free PMC article.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
