

Normalization of hepatic ChREBP activity does not protect against liver disease progression in a mouse model for Glycogen Storage Disease type Ia
- PMID: 37085901
- PMCID: PMC10122297
- DOI: 10.1186/s40170-023-00305-3
Normalization of hepatic ChREBP activity does not protect against liver disease progression in a mouse model for Glycogen Storage Disease type Ia
Abstract
Background: Glycogen storage disease type 1a (GSD Ia) is an inborn error of metabolism caused by a defect in glucose-6-phosphatase (G6PC1) activity, which induces severe hepatomegaly and increases the risk for liver cancer. Hepatic GSD Ia is characterized by constitutive activation of Carbohydrate Response Element Binding Protein (ChREBP), a glucose-sensitive transcription factor. Previously, we showed that ChREBP activation limits non-alcoholic fatty liver disease (NAFLD) in hepatic GSD Ia. As ChREBP has been proposed as a pro-oncogenic molecular switch that supports tumour progression, we hypothesized that ChREBP normalization protects against liver disease progression in hepatic GSD Ia.
Methods: Hepatocyte-specific G6pc knockout (L-G6pc-/-) mice were treated with AAV-shChREBP to normalize hepatic ChREBP activity.
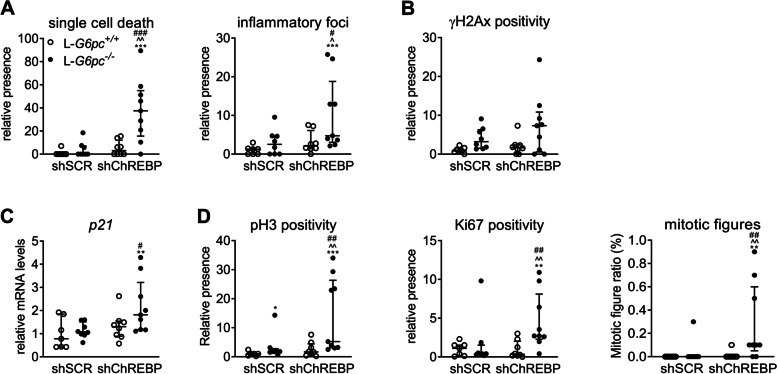
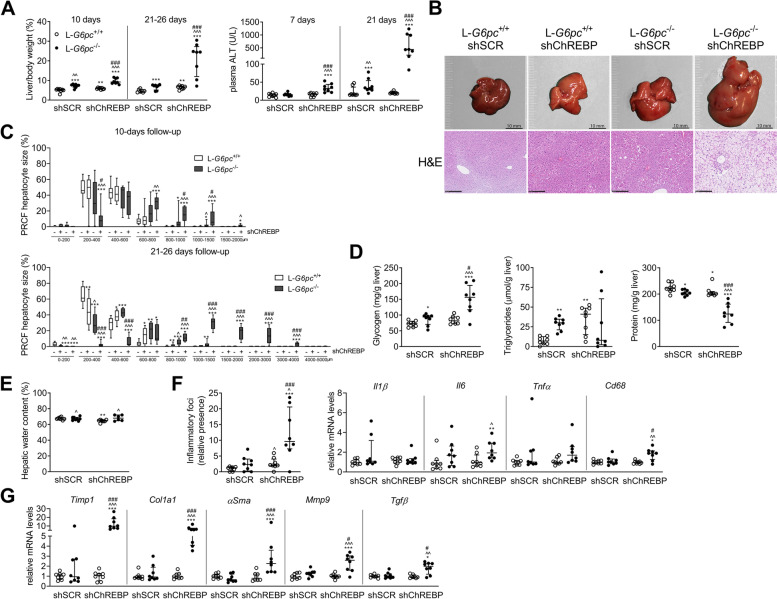
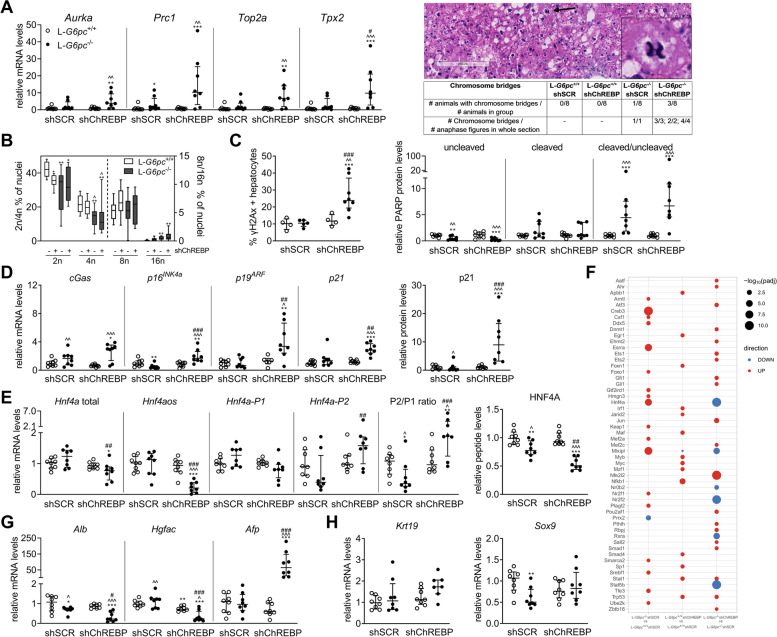
Results: Hepatic ChREBP normalization in GSD Ia mice induced dysplastic liver growth, massively increased hepatocyte size, and was associated with increased hepatic inflammation. Furthermore, nuclear levels of the oncoprotein Yes Associated Protein (YAP) were increased and its transcriptional targets were induced in ChREBP-normalized GSD Ia mice. Hepatic ChREBP normalization furthermore induced DNA damage and mitotic activity in GSD Ia mice, while gene signatures of chromosomal instability, the cytosolic DNA-sensing cGAS-STING pathway, senescence, and hepatocyte dedifferentiation emerged.
Conclusions: In conclusion, our findings indicate that ChREBP activity limits hepatomegaly while decelerating liver disease progression and protecting against chromosomal instability in hepatic GSD Ia. These results disqualify ChREBP as a therapeutic target for treatment of liver disease in GSD Ia. In addition, they underline the importance of establishing the context-specific roles of hepatic ChREBP to define its therapeutic potential to prevent or treat advanced liver disease.
Keywords: Carbohydrate Response Element Binding Protein; Cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING); Glycogen Storage Disease type 1a; Hepatomegaly; Yes Associated Protein.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Rake J, Visser G, Labrune P, Leonard J, Ullrich K, Smit P. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 2002;161 Suppl:S20–34. 10.1007/S00431-002-0999-4. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
