

Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics
- PMID: 37088806
- PMCID: PMC10123065
- DOI: 10.1038/s41408-023-00830-w
Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics
Erratum in
-
Correction: Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics.Blood Cancer J. 2023 May 17;13(1):80. doi: 10.1038/s41408-023-00857-z. Blood Cancer J. 2023. PMID: 37193700 Free PMC article. No abstract available.
Abstract
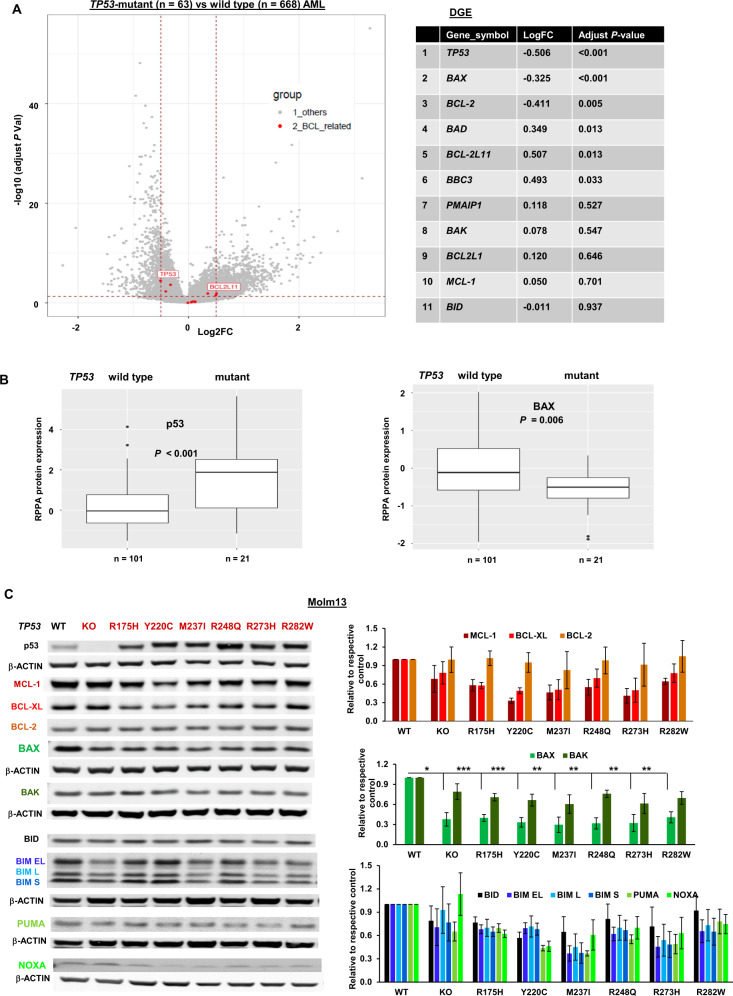
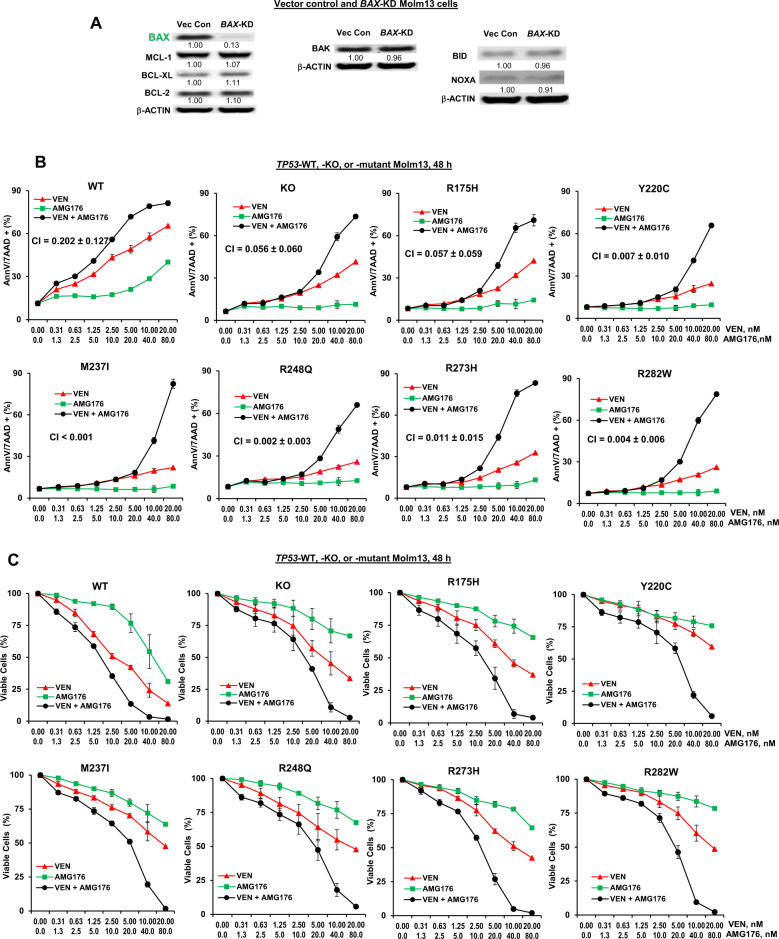
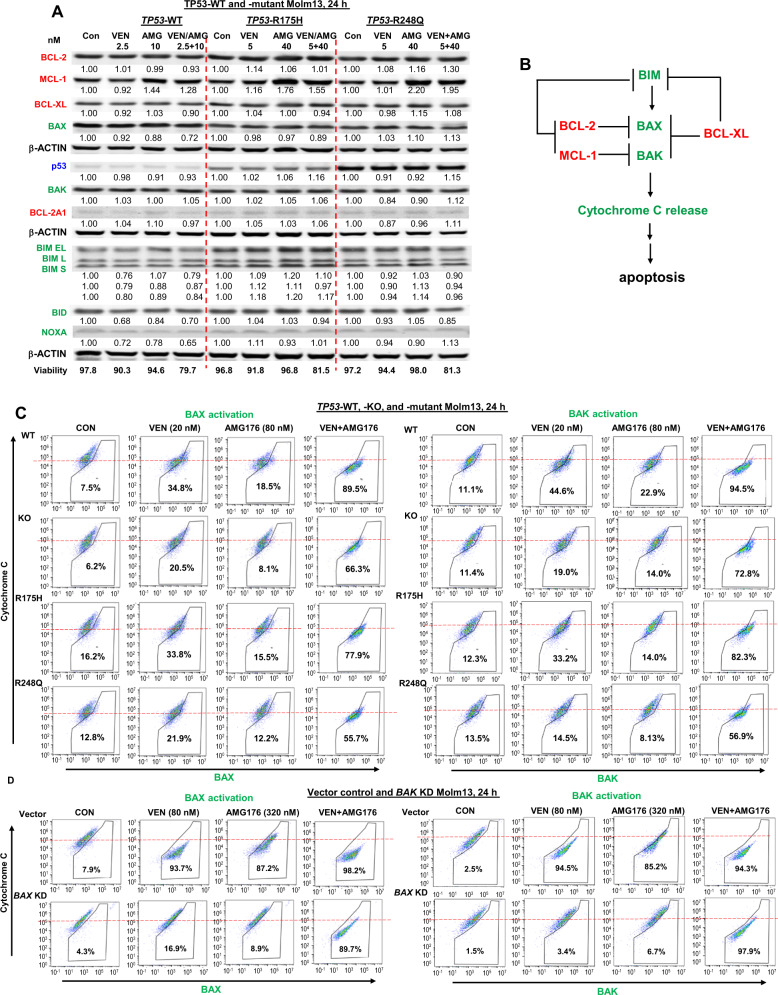
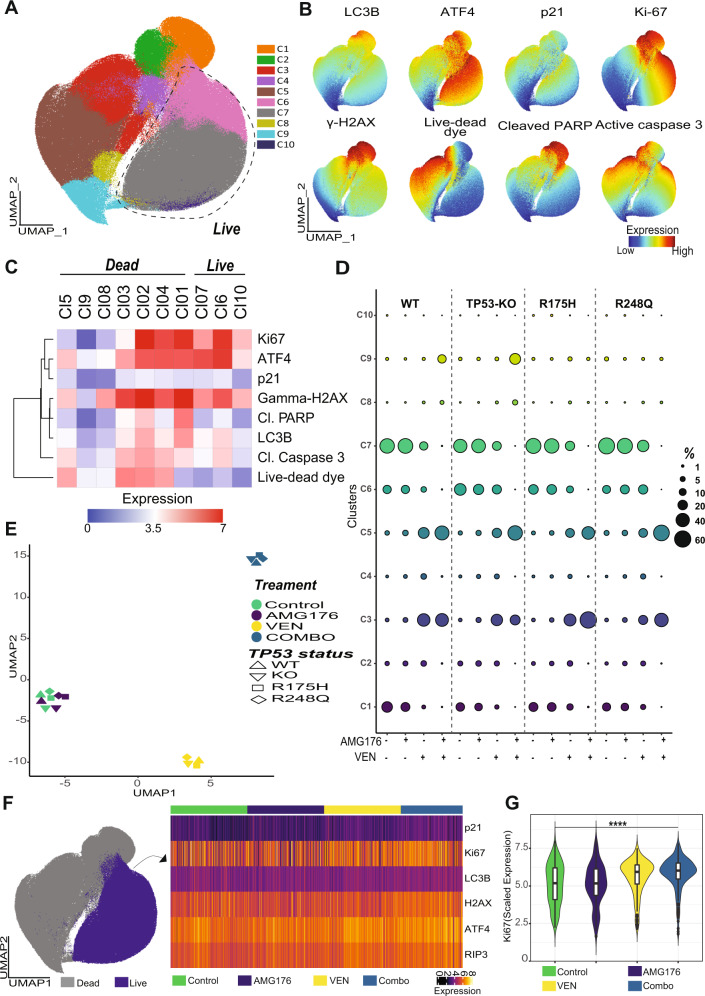
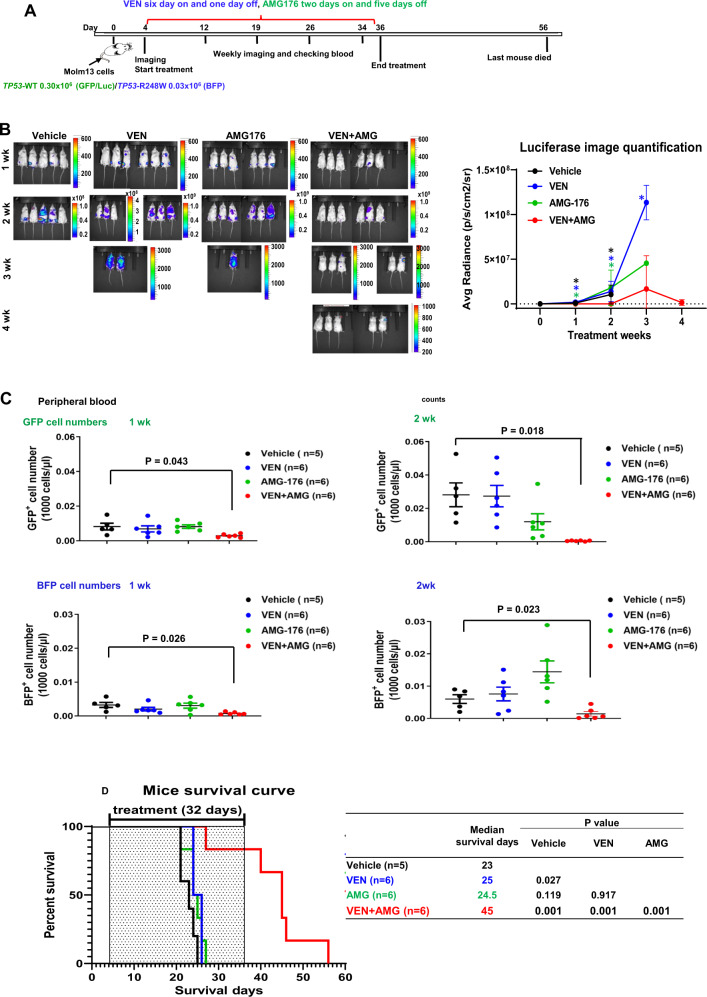
TP53-mutant acute myeloid leukemia (AML) respond poorly to currently available treatments, including venetoclax-based drug combinations and pose a major therapeutic challenge. Analyses of RNA sequencing and reverse phase protein array datasets revealed significantly lower BAX RNA and protein levels in TP53-mutant compared to TP53-wild-type (WT) AML, a finding confirmed in isogenic CRISPR-generated TP53-knockout and -mutant AML. The response to either BCL-2 (venetoclax) or MCL-1 (AMG176) inhibition was BAX-dependent and much reduced in TP53-mutant compared to TP53-WT cells, while the combination of two BH3 mimetics effectively activated BAX, circumventing survival mechanisms in cells treated with either BH3 mimetic, and synergistically induced cell death in TP53-mutant AML and stem/progenitor cells. The BH3 mimetic-driven stress response and cell death patterns after dual inhibition were largely independent of TP53 status and affected by apoptosis induction. Co-targeting, but not individual targeting of BCL-2 and MCL-1 in mice xenografted with TP53-WT and TP53-R248W Molm13 cells suppressed both TP53-WT and TP53-mutant cell growth and significantly prolonged survival. Our results demonstrate that co-targeting BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance to individual BH3 mimetics in TP53-mutant cells, thus shifting cell fate from survival to death in TP53-deficient and -mutant AML. This concept warrants clinical evaluation.
© 2023. The Author(s).
Conflict of interest statement
PEH and PKM are employees of Amgen.
Figures

References
-
- Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19:1405–13. doi: 10.1200/JCO.2001.19.5.1405. - DOI - PubMed
-
- Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39:348–54. doi: 10.1016/j.leukres.2014.12.006. - DOI - PMC - PubMed
-
- Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–21. doi: 10.1182/blood-2011-08-375758. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
