

Effects of Selection at Linked Sites on Patterns of Genetic Variability
- PMID: 37089401
- PMCID: PMC10120885
- DOI: 10.1146/annurev-ecolsys-010621-044528
Effects of Selection at Linked Sites on Patterns of Genetic Variability
Abstract
Patterns of variation and evolution at a given site in a genome can be strongly influenced by the effects of selection at genetically linked sites. In particular, the recombination rates of genomic regions correlate with their amount of within-population genetic variability, the degree to which the frequency distributions of DNA sequence variants differ from their neutral expectations, and the levels of adaptation of their functional components. We review the major population genetic processes that are thought to lead to these patterns, focusing on their effects on patterns of variability: selective sweeps, background selection, associative overdominance, and Hill-Robertson interference among deleterious mutations. We emphasize the difficulties in distinguishing among the footprints of these processes and disentangling them from the effects of purely demographic factors such as population size changes. We also discuss how interactions between selective and demographic processes can significantly affect patterns of variability within genomes.
Keywords: Hill–Robertson interference; associative overdominance; background selection; genetic recombination; hitchhiking; selective sweeps.
Figures
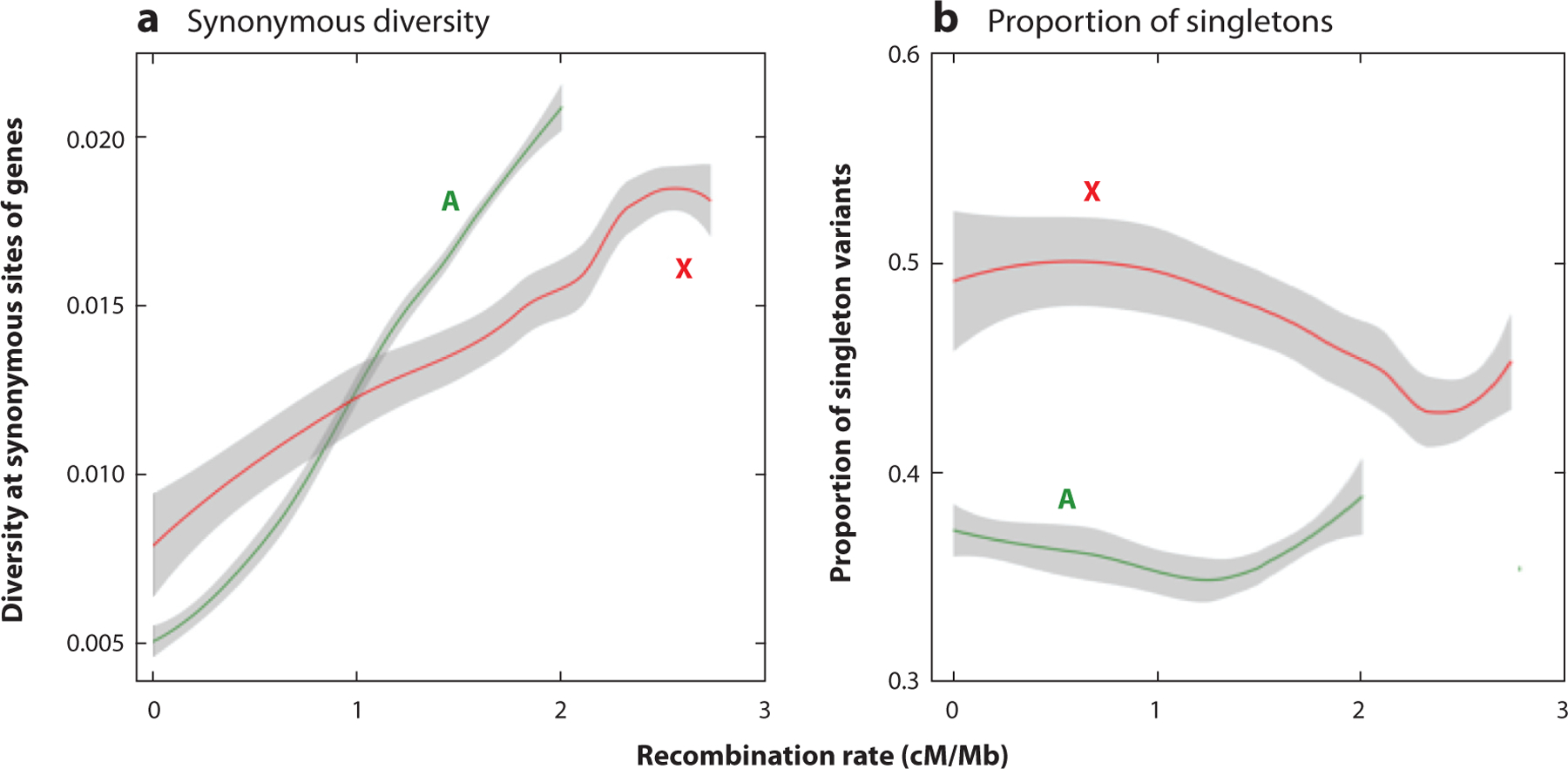
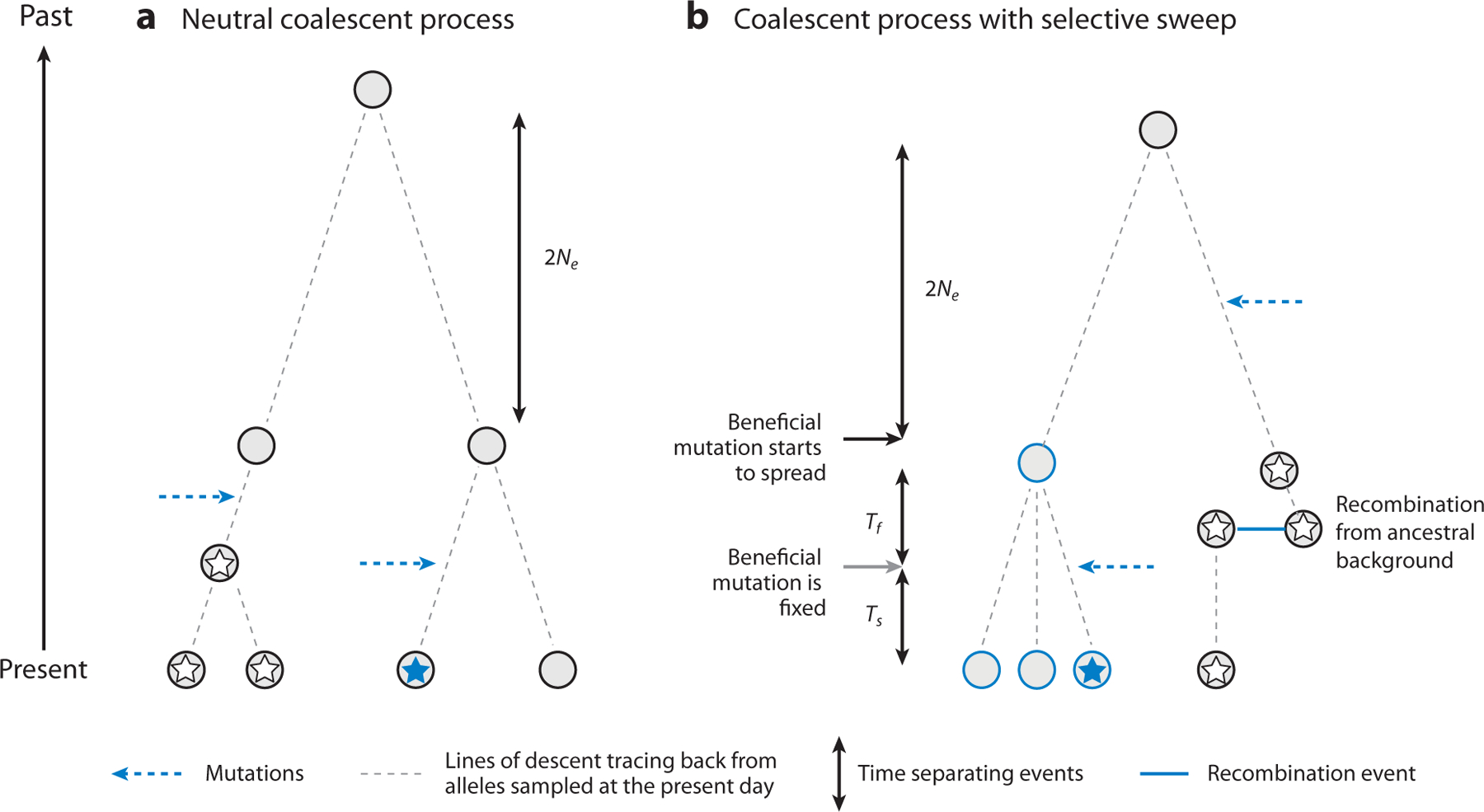
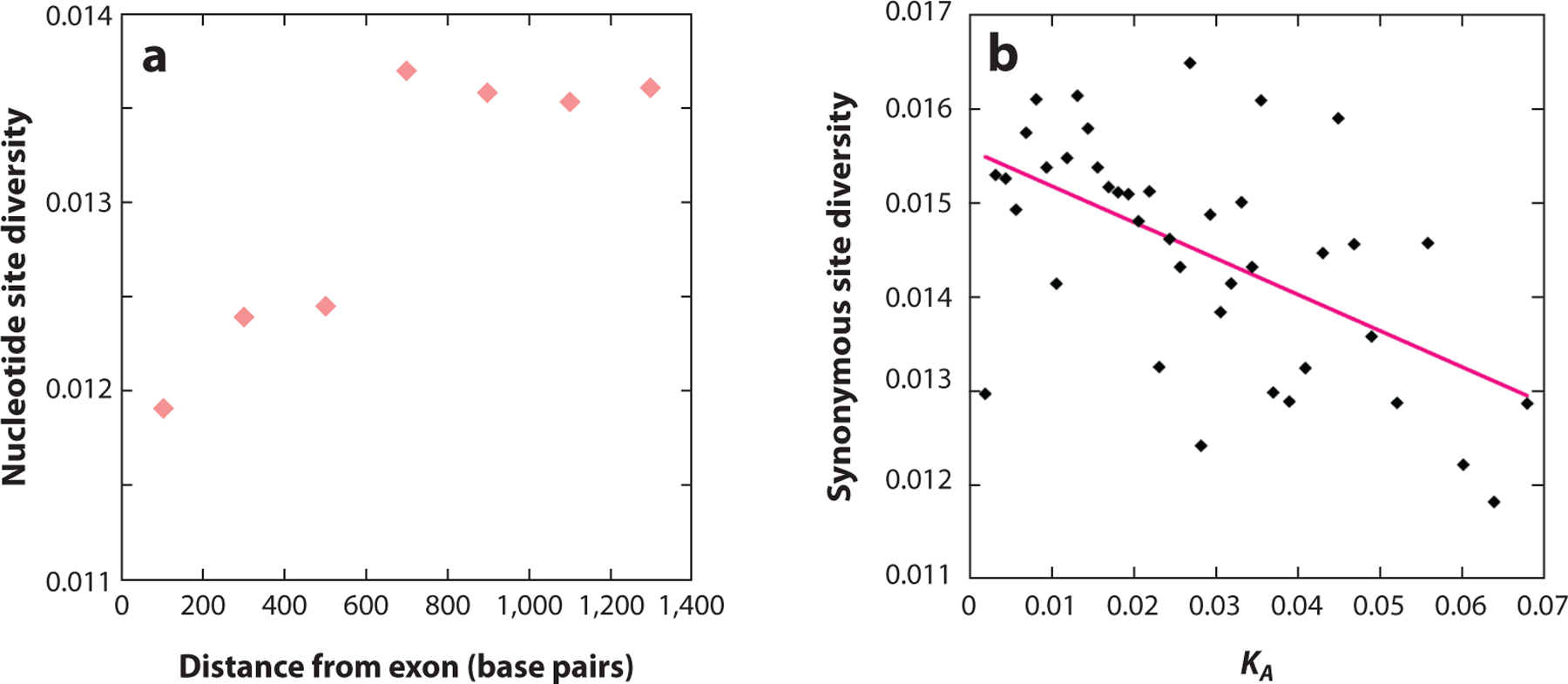
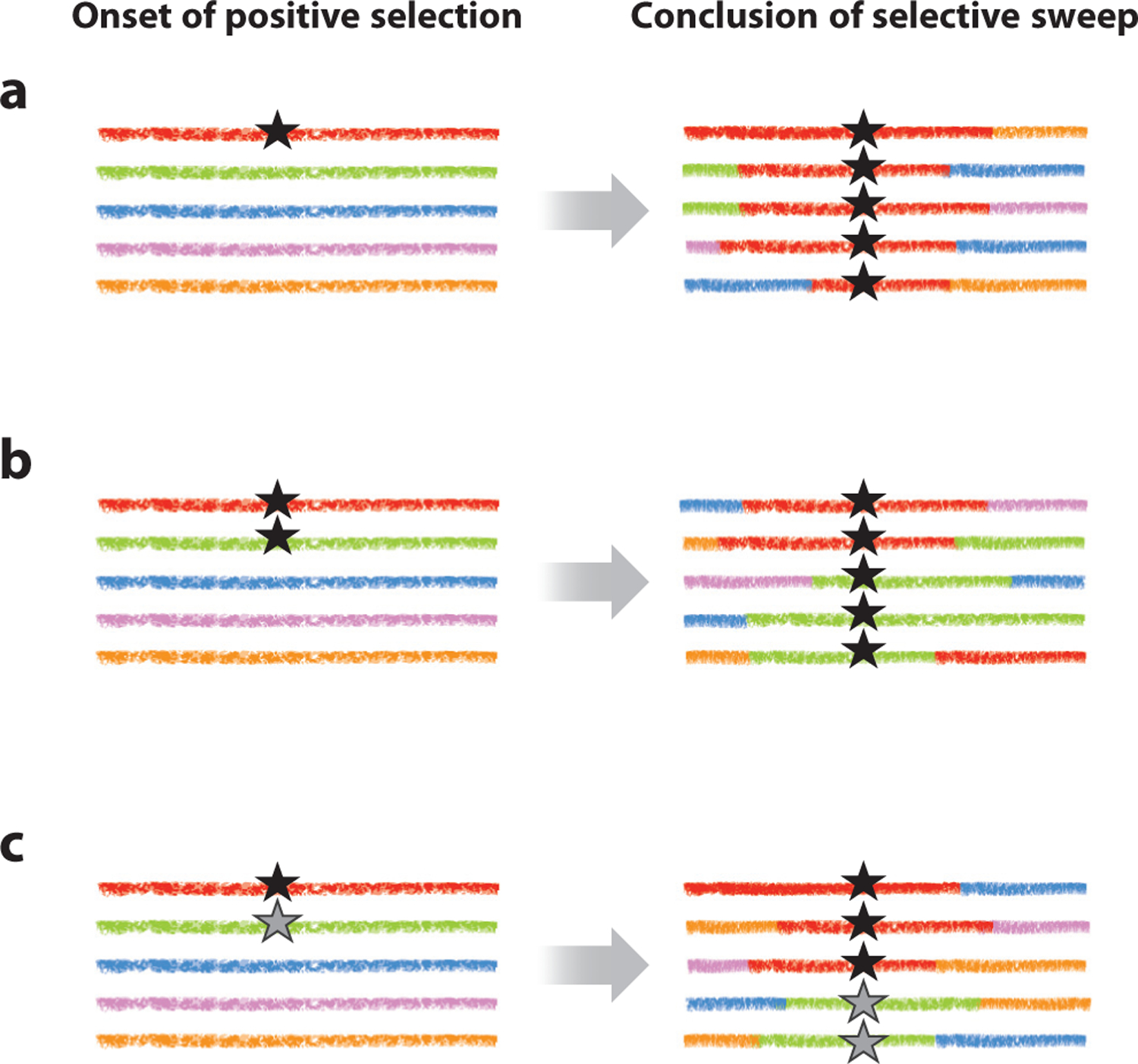
References
-
- Bank C, Foll M, Ferrer-Admetlla A, Ewing G, Jensen JD. 2014. Thinking too positive? Revisiting current methods in population genetic selection inference. Trends Genet 30:540–46 - PubMed
-
- Barton NH. 1979. Gene flow past a cline. Heredity 43:333–39
-
- Becher H, Jackson BC, Charlesworth B. 2020. Patterns of genetic variability in genomic regions with low rates of recombination. Curr. Biol 30:94–100 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources