

Interactions between carbon nanotubes and external structures of SARS-CoV-2 using molecular docking and molecular dynamics
- PMID: 37089815
- PMCID: PMC10111146
- DOI: 10.1016/j.molstruc.2023.135604
Interactions between carbon nanotubes and external structures of SARS-CoV-2 using molecular docking and molecular dynamics
Abstract
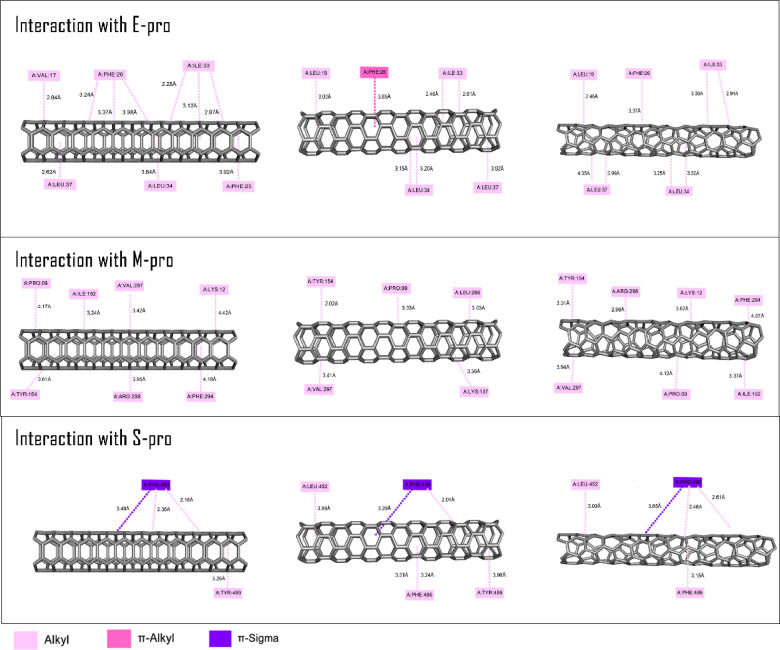
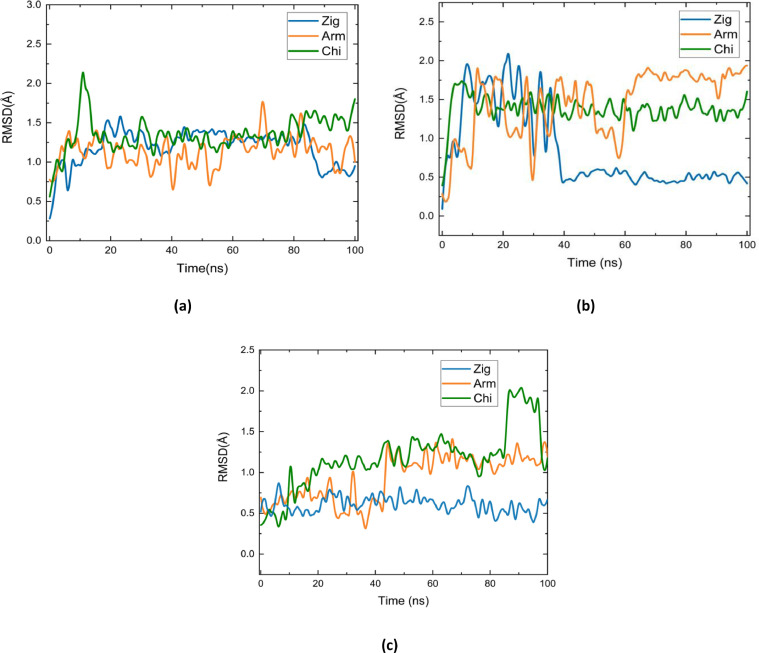
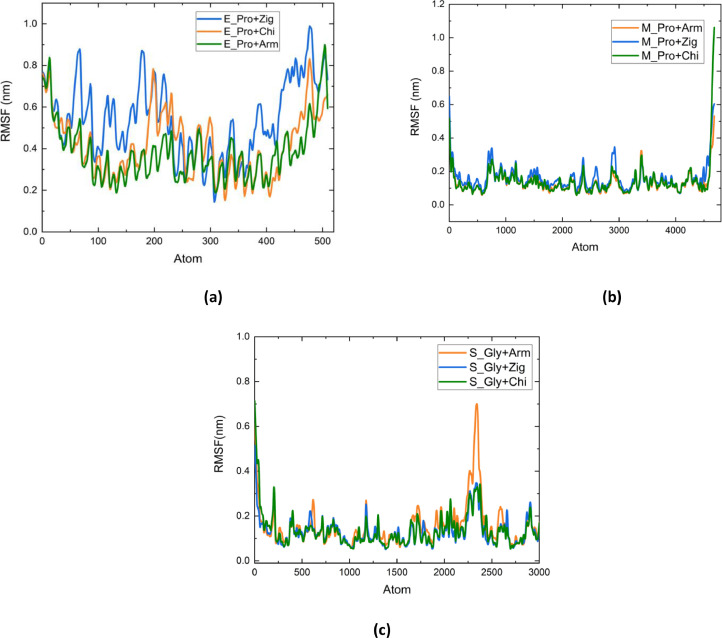
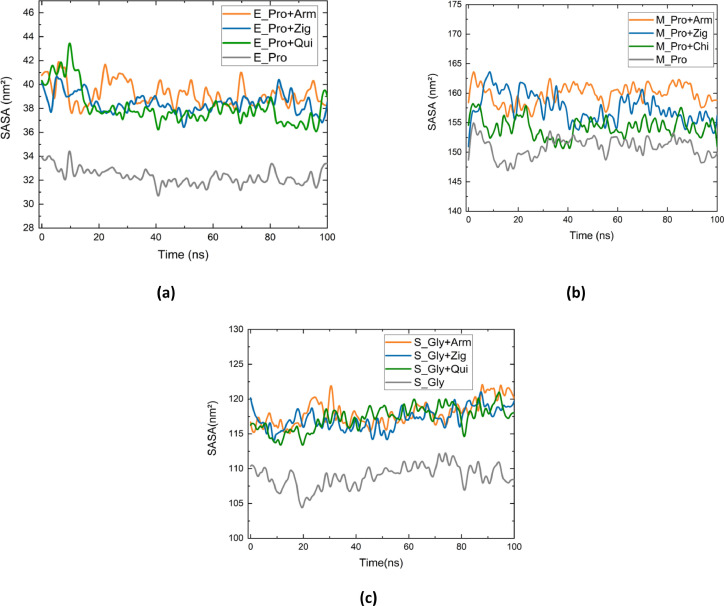
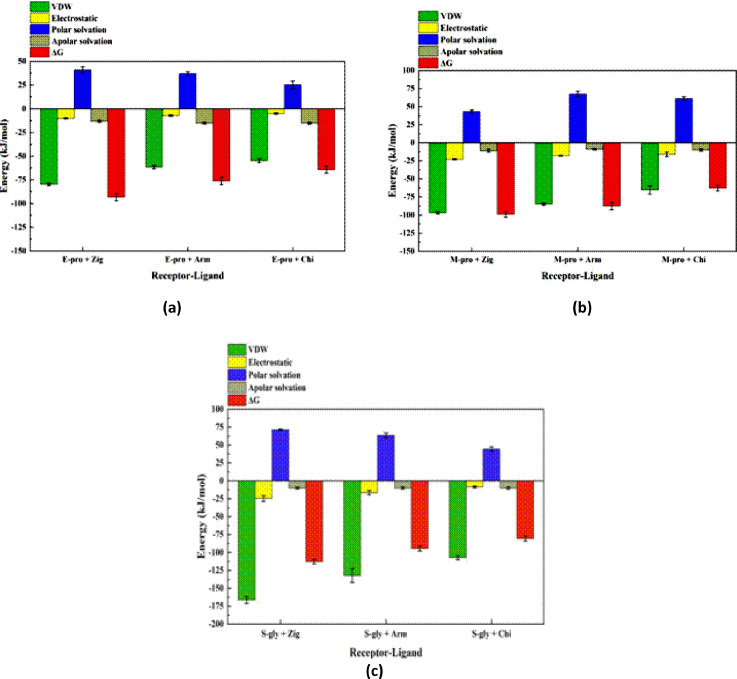
Molecular modeling techniques are used to describe the process of interaction between nanotubes and the main structures of the Covid-19 virus: the envelope protein, the main protease, and the Spike glycoprotein. Molecular docking studies show that the ligands have interaction characteristics capable of adsorbing the structures. Molecular dynamics simulations provide information on the mean squared deviation of atomic positions between 0.5 and 3.0 Å. The Gibbs free energy model and solvent accessible surface area approaches are used. Through the results obtained through molecular dynamics simulations, it is noted that the zig-zag nanotube prefers to interact with E-pro, M-pro, and S-gly, respectively. Molecular couplings and free energy showed that the S-gly active site residues strongly interact with zigzag, chiral, and armchair nanotubes, in this order. The interactions demonstrated in this manuscript may predict some promising candidates for virus antagonists, which may be confirmed through experimental approaches.
Keywords: Antiviral effect; Carbon nanotube; Molecular docking, Molecular dynamics, DFT, In silico study; SARS–CoV-2.
© 2023 Published by Elsevier B.V.
Conflict of interest statement
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Figures

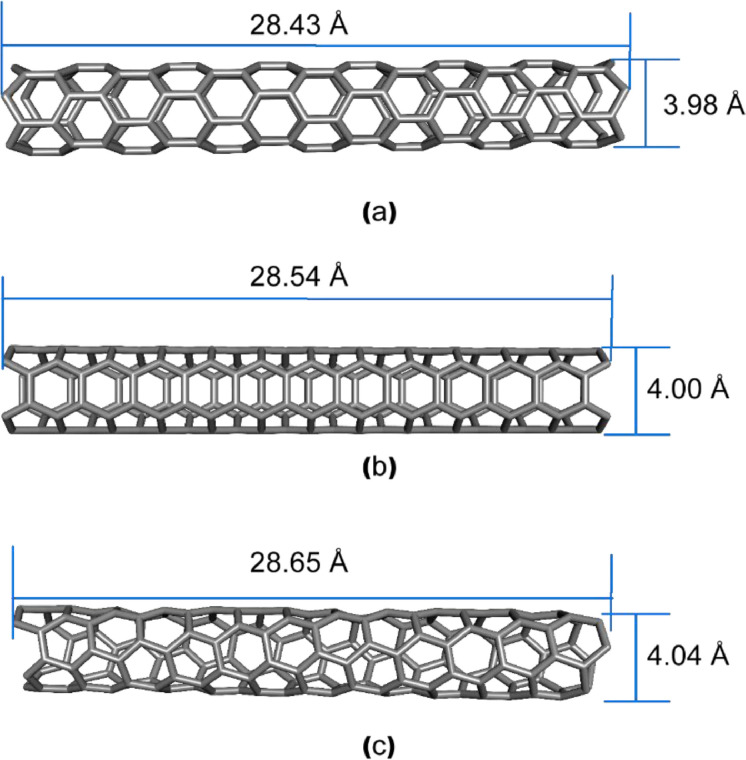
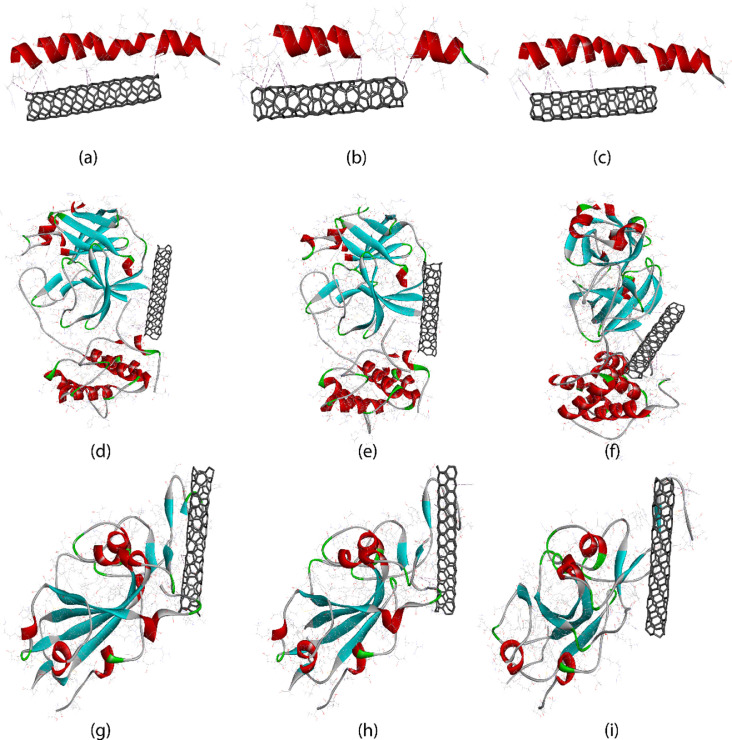
References
-
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindah E. Gromacs: high-performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. - DOI
-
- Arouche T.da S., Reis A.F., Martins A.Y., Costa J.F.S., Carvalho Junior R.N., Neto J.C., A M. Interactions between remdesivir, ribavirin, favipiravir, galidesivir, hydroxychloroquine and chloroquine with fragment molecular of the COVID-19 main protease with inhibitor N3 complex (PDB ID:6LU7) using molecular docking. J. Nanosci. Nanotechnol. 2020;20(12):7311–7323. doi: 10.1166/jnn.2020.18955. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
