

Towards mechanistic models of mutational effects: Deep learning on Alzheimer's Aβ peptide
- PMID: 37090430
- PMCID: PMC10114515
- DOI: 10.1016/j.csbj.2023.03.051
Towards mechanistic models of mutational effects: Deep learning on Alzheimer's Aβ peptide
Abstract
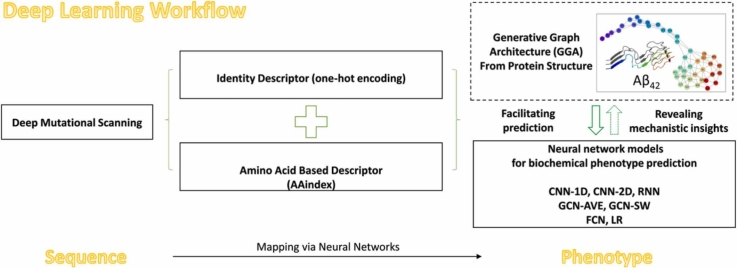
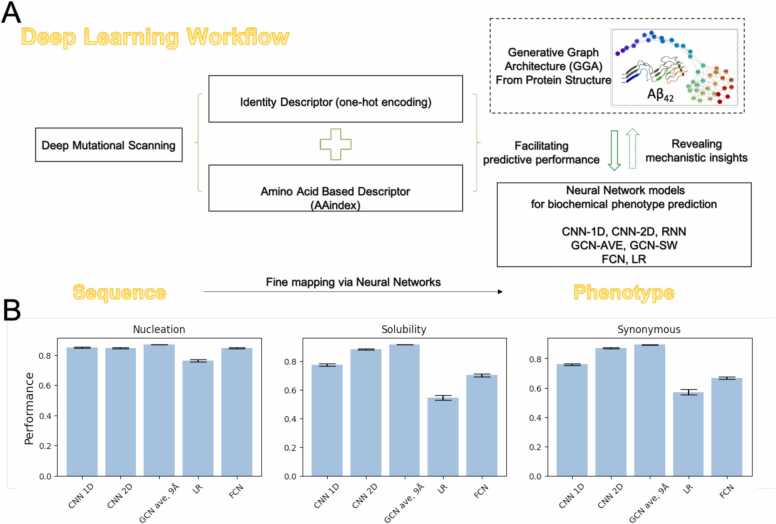
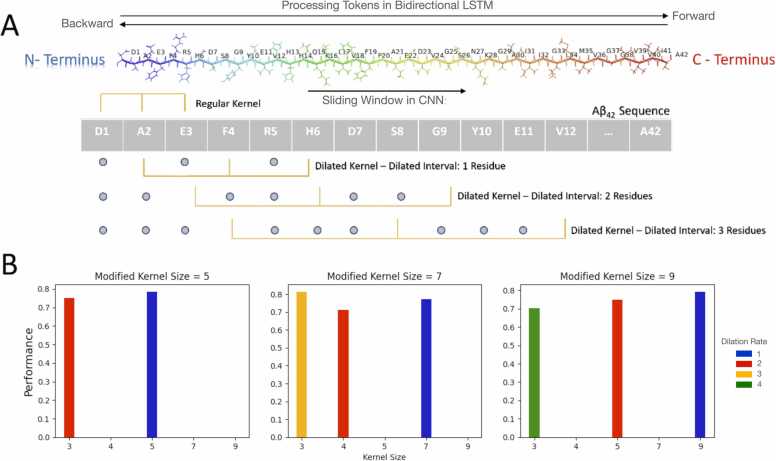
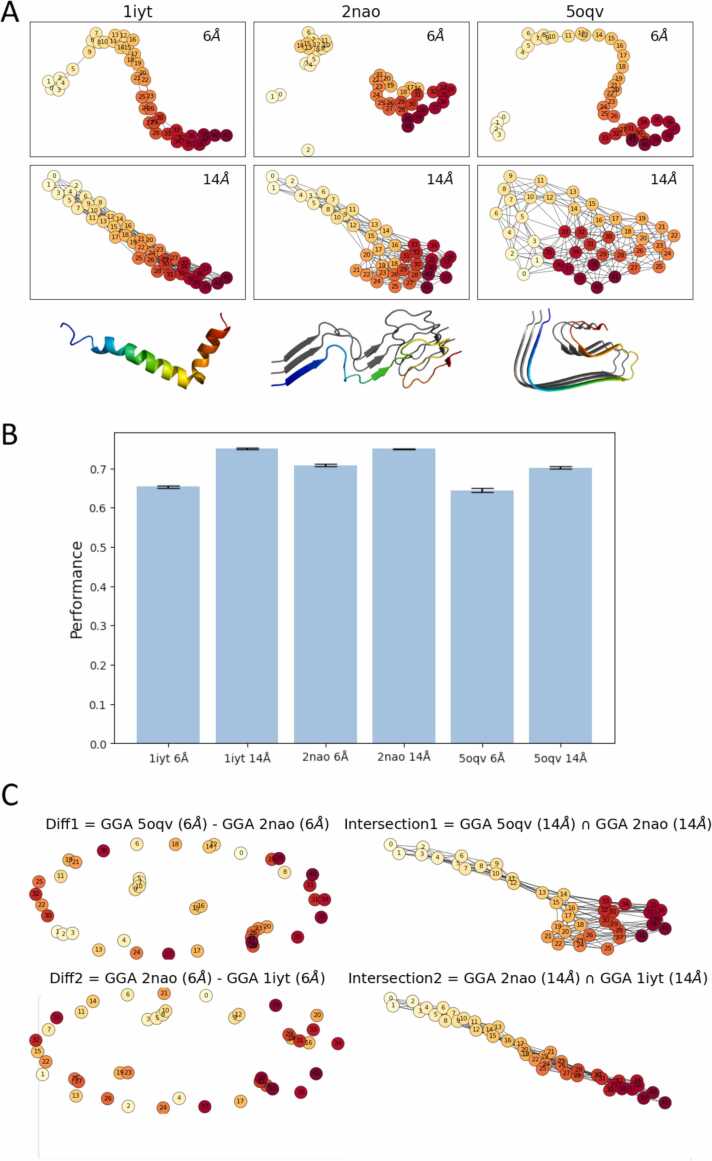
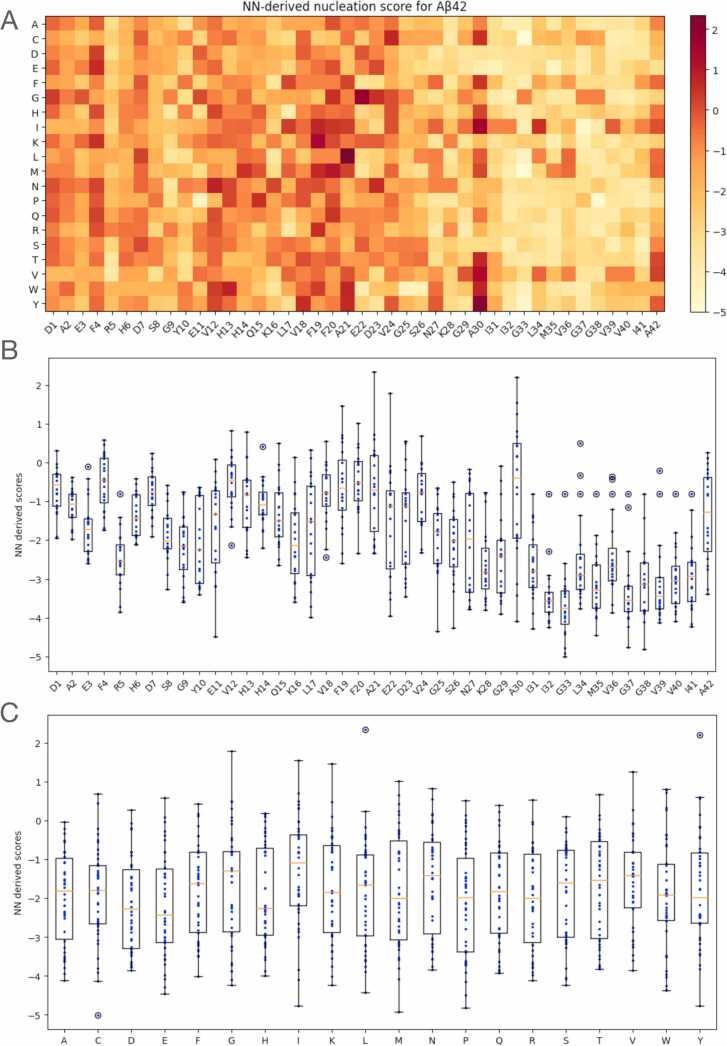
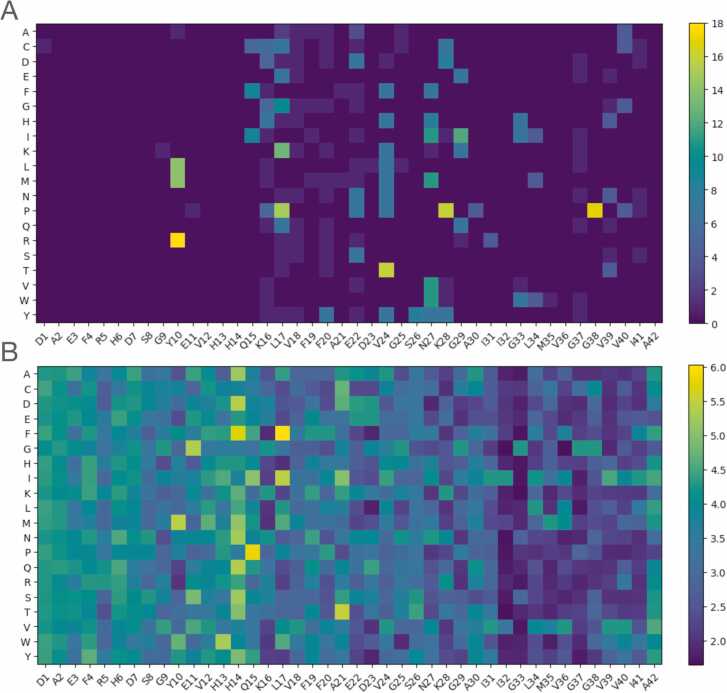
Deep Mutational Scanning (DMS) has enabled multiplexed measurement of mutational effects on protein properties, including kinematics and self-organization, with unprecedented resolution. However, potential bottlenecks of DMS characterization include experimental design, data quality, and depth of mutational coverage. Here, we apply deep learning to comprehensively model the mutational effect of the Alzheimer's Disease associated peptide Aβ42 on aggregation-related biochemical traits from DMS measurements. Among tested neural network architectures, Convolutional Neural Networks and Recurrent Neural Networks are found to be the most cost-effective models with high performance even under insufficiently-sampled DMS studies. While sequence features are essential for satisfactory prediction from neural networks, geometric-structural features further enhance the prediction performance. Notably, we demonstrate how mechanistic insights into phenotype may be extracted from the neural networks themselves suitably designed. This methodological benefit is particularly relevant for biochemical systems displaying a strong coupling between structure and phenotype such as the conformation of Aβ42 aggregate and nucleation, as shown here using a Graph Convolutional Neural Network (GCN) developed from the protein atomic structure input. In addition to accurate imputation of missing values (which here ranged up to 55% of all phenotype values at key residues), the mutationally-defined nucleation phenotype generated from a GCN shows improved resolution for identifying known disease-causing mutations relative to the original DMS phenotype. Our study suggests that neural network derived sequence-phenotype mapping can be exploited not only to provide direct support for protein engineering or genome editing but also to facilitate therapeutic design with the gained perspectives from biological modeling.
Keywords: Alzheimer's disease; Convolutional neural networks; Deep learning; Deep mutational scanning; Mutation; Neural networks; Nucleation; Recurrent neural networks.
© 2023 The Author(s).
Conflict of interest statement
Eric R. Gamazon receives an honorarium from the journal Circulation Research of the American Heart Association, as a member of the Editorial Board.
Figures
References
-
- Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
LinkOut - more resources
Full Text Sources