

CDEMI: Characterizing differences in microbial composition and function in microbiome data
- PMID: 37090432
- PMCID: PMC10113763
- DOI: 10.1016/j.csbj.2023.03.044
CDEMI: Characterizing differences in microbial composition and function in microbiome data
Abstract
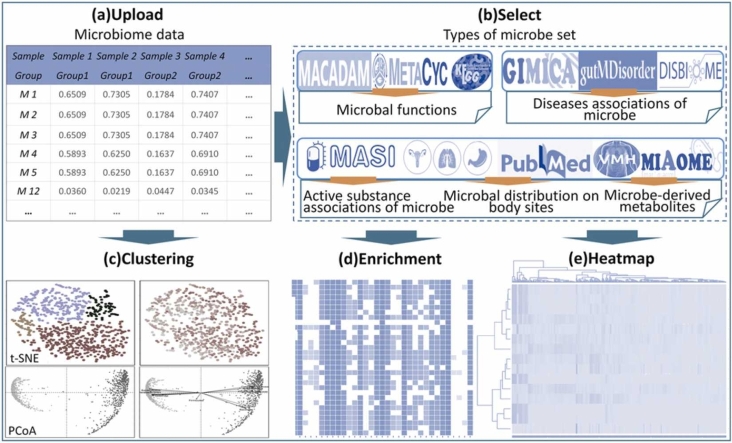
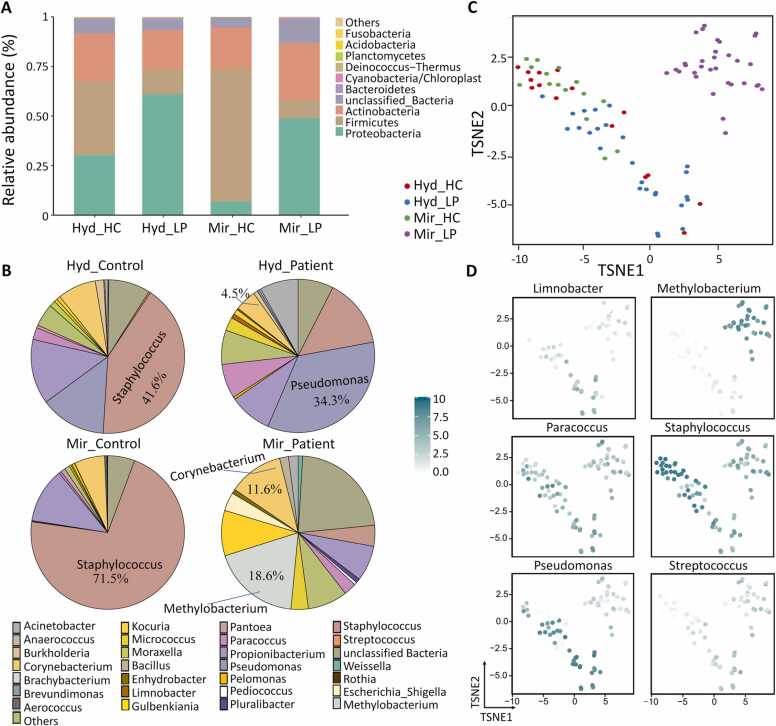
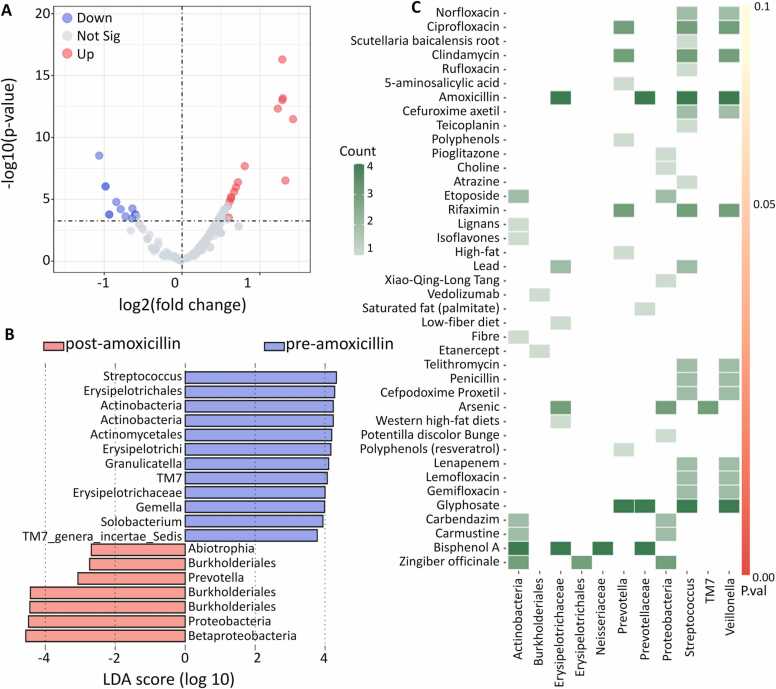
Microbial communities influence host phenotypes through microbiota-derived metabolites and interactions between exogenous active substances (EASs) and the microbiota. Owing to the high dynamics of microbial community composition and difficulty in microbial functional analysis, the identification of mechanistic links between individual microbes and host phenotypes is complex. Thus, it is important to characterize variations in microbial composition across various conditions (for example, topographical locations, times, physiological and pathological conditions, and populations of different ethnicities) in microbiome studies. However, no web server is currently available to facilitate such characterization. Moreover, accurately annotating the functions of microbes and investigating the possible factors that shape microbial function are critical for discovering links between microbes and host phenotypes. Herein, an online tool, CDEMI, is introduced to discover microbial composition variations across different conditions, and five types of microbe libraries are provided to comprehensively characterize the functionality of microbes from different perspectives. These collective microbe libraries include (1) microbial functional pathways, (2) disease associations with microbes, (3) EASs associations with microbes, (4) bioactive microbial metabolites, and (5) human body habitats. In summary, CDEMI is unique in that it can reveal microbial patterns in distributions/compositions across different conditions and facilitate biological interpretations based on diverse microbe libraries. CDEMI is accessible at http://rdblab.cn/cdemi/.
Keywords: Functional characterization; Metabolic pathway; Microbial association; Microbial composition; Microbiome.
© 2023 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures



Similar articles
-
Mechanistic links between gut microbial community dynamics, microbial functions and metabolic health.World J Gastroenterol. 2014 Nov 28;20(44):16498-517. doi: 10.3748/wjg.v20.i44.16498. World J Gastroenterol. 2014. PMID: 25469018 Free PMC article. Review.
-
The Age of Next-Generation Therapeutic-Microbe Discovery: Exploiting Microbe-Microbe and Host-Microbe Interactions for Disease Prevention.Infect Immun. 2022 May 19;90(5):e0058921. doi: 10.1128/iai.00589-21. Epub 2022 Apr 6. Infect Immun. 2022. PMID: 35384688 Free PMC article. Review.
-
Microbial Hub Taxa Link Host and Abiotic Factors to Plant Microbiome Variation.PLoS Biol. 2016 Jan 20;14(1):e1002352. doi: 10.1371/journal.pbio.1002352. eCollection 2016 Jan. PLoS Biol. 2016. PMID: 26788878 Free PMC article.
-
Microbial Turnover and Dispersal Events Occur in Synchrony with Plant Phenology in the Perennial Evergreen Tree Crop Citrus sinensis.mBio. 2022 Jun 28;13(3):e0034322. doi: 10.1128/mbio.00343-22. Epub 2022 Jun 1. mBio. 2022. PMID: 35642946 Free PMC article.
-
MicroPattern: a web-based tool for microbe set enrichment analysis and disease similarity calculation based on a list of microbes.Sci Rep. 2017 Jan 10;7:40200. doi: 10.1038/srep40200. Sci Rep. 2017. PMID: 28071710 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases