

This is a preprint.
SMYD2 Regulates Vascular Smooth Muscle Cell Phenotypic Switching and Intimal Hyperplasia via Interaction with Myocardin
- PMID: 37090651
- PMCID: PMC10120764
- DOI: 10.21203/rs.3.rs-2721176/v1
SMYD2 Regulates Vascular Smooth Muscle Cell Phenotypic Switching and Intimal Hyperplasia via Interaction with Myocardin
Update in
-
SMYD2 regulates vascular smooth muscle cell phenotypic switching and intimal hyperplasia via interaction with myocardin.Cell Mol Life Sci. 2023 Aug 24;80(9):264. doi: 10.1007/s00018-023-04883-9. Cell Mol Life Sci. 2023. PMID: 37615725 Free PMC article.
Abstract
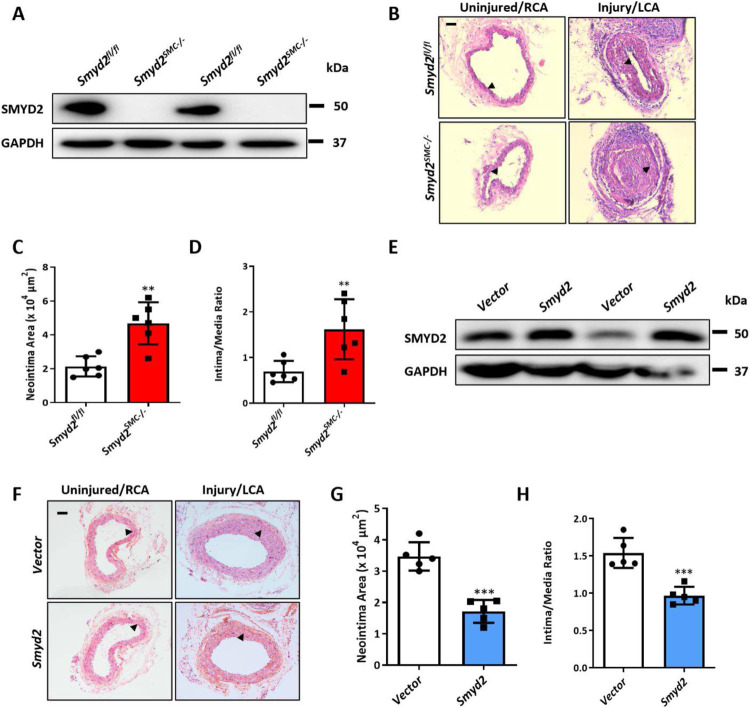
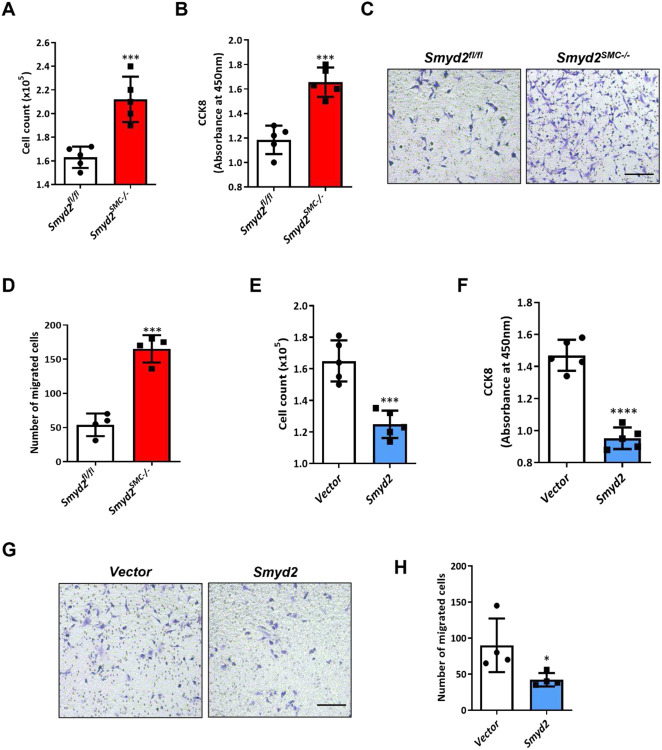
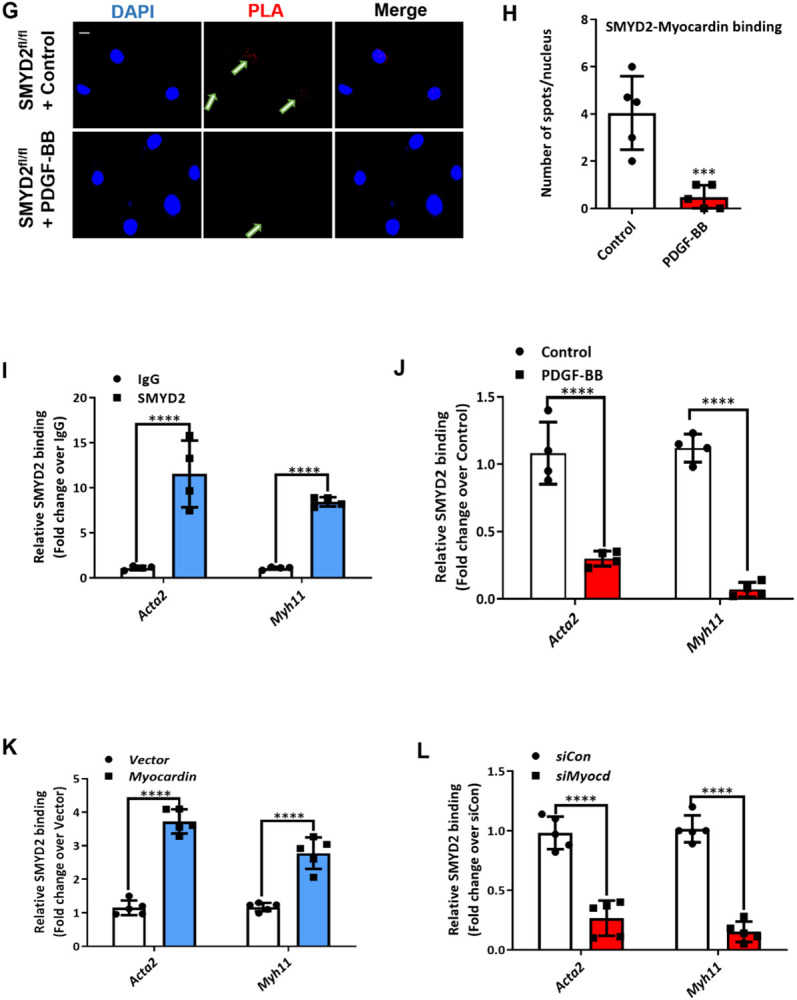
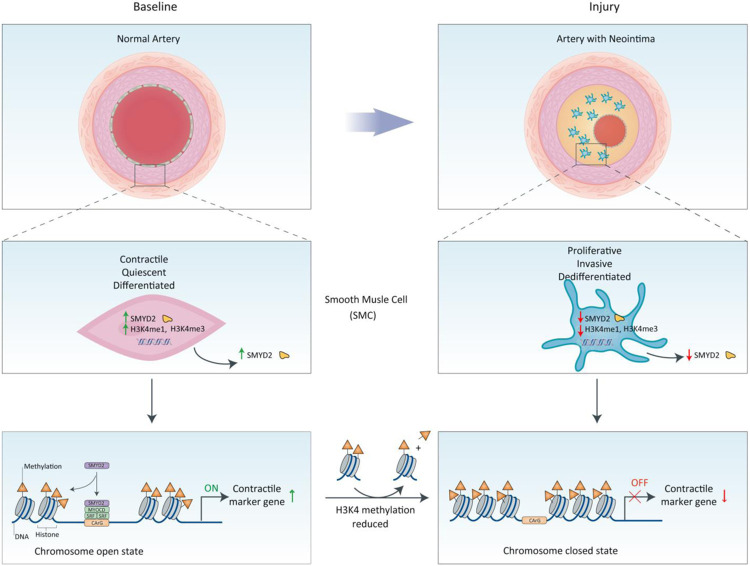
The SET and MYND domain-containing protein 2 (SMYD2) is a histone lysine methyltransferase that has been reported to regulate carcinogenesis and inflammation. However, its role in vascular smooth muscle cell (VSMC) homeostasis and vascular diseases has not been determined. Here, we investigated the role of SMYD2 in VSMC phenotypic modulation and vascular intimal hyperplasia and elucidated the underlying mechanism. We observed that SMYD2 expression was downregulated in injured carotid arteries in mice and phenotypically modulated VSMCs in vitro. Using a SMC-specific Smyd2 knockout mouse model, we found that Smyd2 ablation in VSMCs exacerbates neointima formation after vascular injury in vivo. Conversely, Smyd2 overexpression inhibits VSMC proliferation and migration in vitro and attenuates arterial narrowing in injured vessels in mice. Smyd2 downregulation promotes VSMC phenotypic switching accompanied with enhanced proliferation and migration. Mechanistically, genome-wide transcriptome analysis and loss/gain-of-function studies revealed that SMYD2 up-regulates VSMC contractile gene expression and suppresses VSMC proliferation and migration, in part, by promoting expression and transactivation of the master transcription cofactor myocardin. In addition, myocardin directly interacts with SMYD2, thereby facilitating SMYD2 recruitment to the CArG regions of SMC contractile gene promoters and leading to an open chromatin status around SMC contractile gene promoters via SMYD2-mediated H3K4 methylation. Hence, we conclude that SMYD2 is a novel regulator of VSMC contractile phenotype and intimal hyperplasia via a myocardin-dependent epigenetic regulatory mechanism and may be a potential therapeutic target for occlusive vascular diseases.
Conflict of interest statement
Conflict of interest
All authors declare no potential conflicts of interest.
Figures





References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
