

This is a preprint.
Evolutionary and immune microenvironment dynamics during neoadjuvant treatment of oesophagael adenocarcinoma
- PMID: 37090678
- PMCID: PMC10120745
- DOI: 10.21203/rs.3.rs-2738048/v1
Evolutionary and immune microenvironment dynamics during neoadjuvant treatment of oesophagael adenocarcinoma
Update in
-
Evolutionary and immune microenvironment dynamics during neoadjuvant treatment of esophageal adenocarcinoma.Nat Cancer. 2025 May;6(5):820-837. doi: 10.1038/s43018-025-00955-w. Epub 2025 May 14. Nat Cancer. 2025. PMID: 40369175 Free PMC article.
Abstract
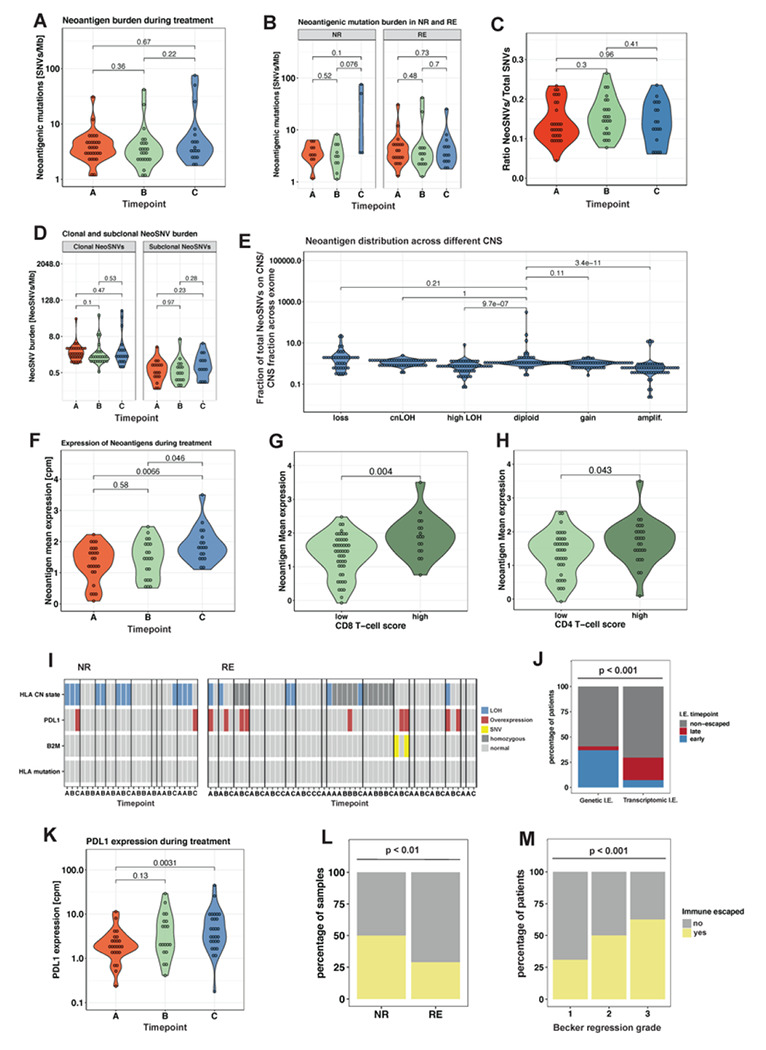
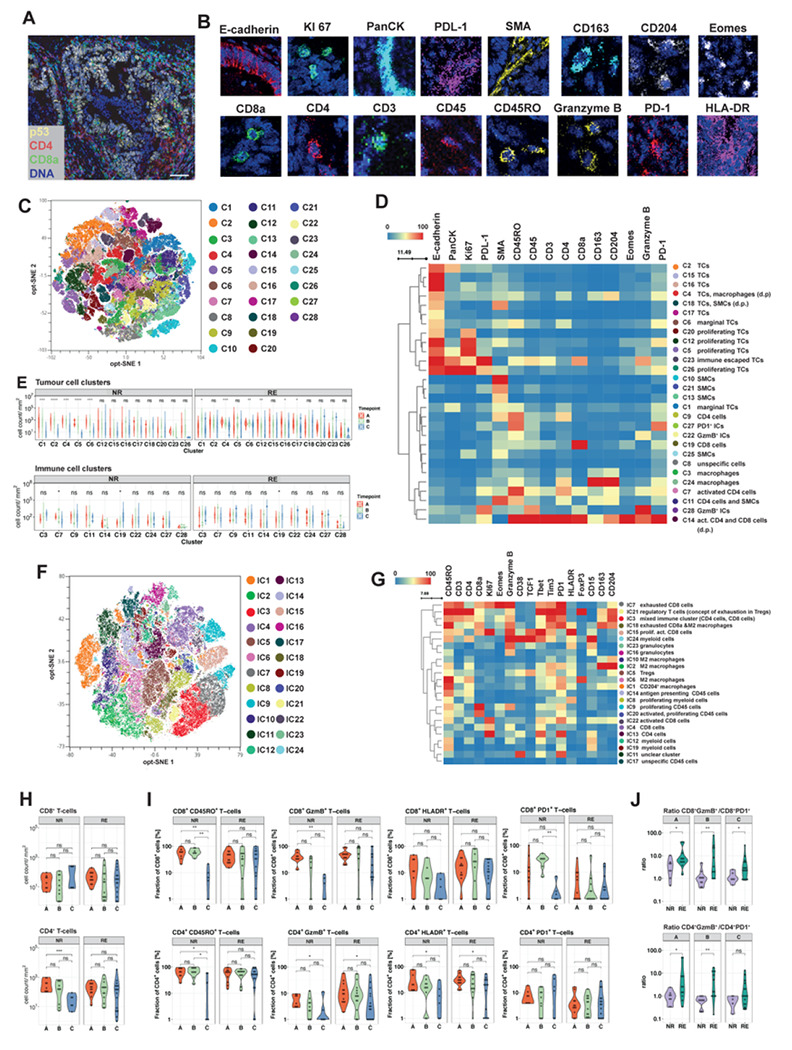
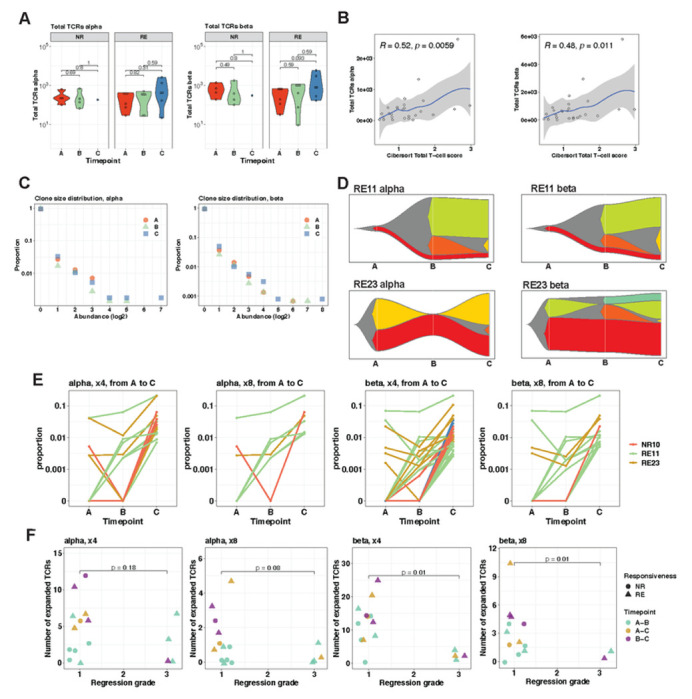
Locally advanced oesophageal adenocarcinoma (EAC) remains difficult to treat because of common resistance to neoadjuvant therapy and high recurrence rates. The ecological and evolutionary dynamics responsible for treatment failure are incompletely understood. Here, we performed a comprehensive multi-omic analysis of samples collected from EAC patients in the MEMORI clinical trial, revealing major changes in gene expression profiles and immune microenvironment composition that did not appear to be driven by changes in clonal composition. Multi-region multi-timepoint whole exome (300x depth) and paired transcriptome sequencing was performed on 27 patients pre-, during and after neoadjuvant treatment. EAC showed major transcriptomic changes during treatment with upregulation of immune and stromal pathways and oncogenic pathways such as KRAS, Hedgehog and WNT. However, genetic data revealed that clonal sweeps were rare, suggesting that gene expression changes were not clonally driven. Additional longitudinal image mass cytometry was performed in a subset of 15 patients and T-cell receptor sequencing in 10 patients, revealing remodelling of the T-cell compartment during treatment and other shifts in microenvironment composition. The presence of immune escape mechanisms and a lack of clonal T-cell expansions were linked to poor clinical treatment response. This study identifies profound transcriptional changes during treatment with limited evidence that clonal replacement is the cause, suggesting phenotypic plasticity and immune dynamics as mechanisms for therapy resistance with pharmacological relevance.
Figures




References
-
- Murugaesu N. et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov 5, 821–831, doi:10.1158/2159-8290.CD-15-0412 (2015). - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
