

Identifying high-confidence variants in human cytomegalovirus genomes sequenced from clinical samples
- PMID: 37091479
- PMCID: PMC10120596
- DOI: 10.1093/ve/veac114
Identifying high-confidence variants in human cytomegalovirus genomes sequenced from clinical samples
Abstract
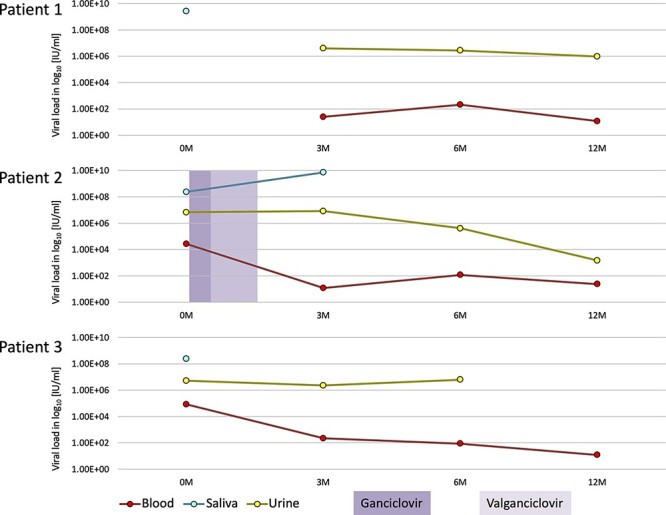
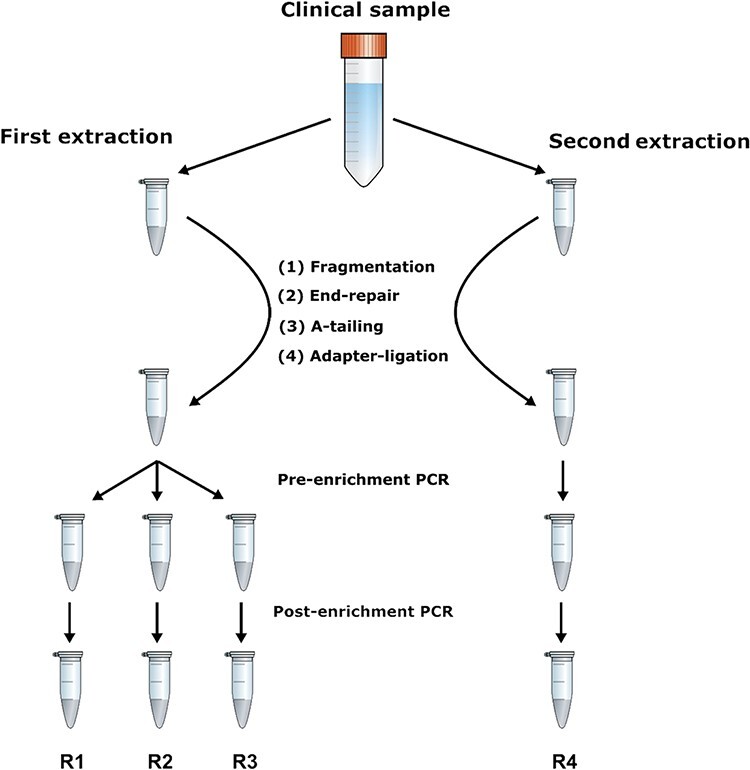
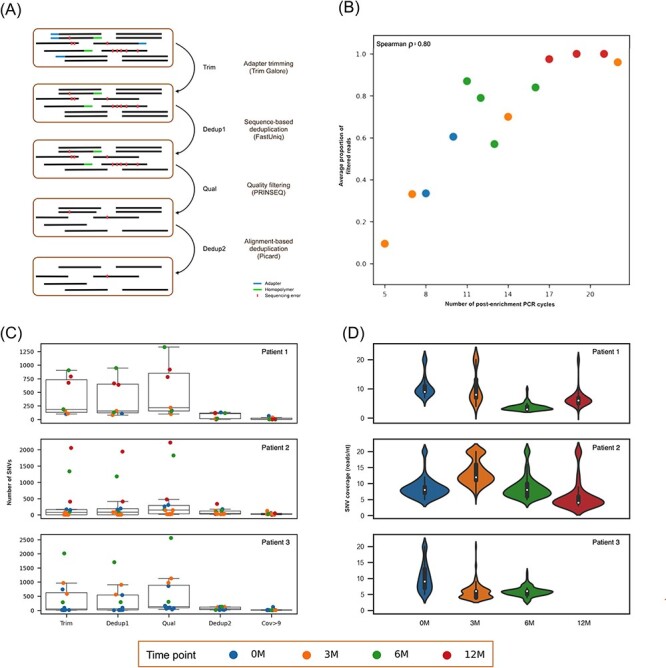
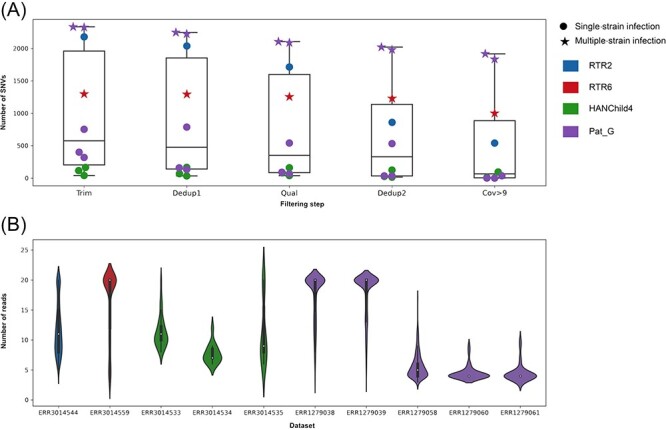
Understanding the intrahost evolution of viral populations has implications in pathogenesis, diagnosis, and treatment and has recently made impressive advances from developments in high-throughput sequencing. However, the underlying analyses are very sensitive to sources of bias, error, and artefact in the data, and it is important that these are addressed adequately if robust conclusions are to be drawn. The key factors include (1) determining the number of viral strains present in the sample analysed; (2) monitoring the extent to which the data represent these strains and assessing the quality of these data; (3) dealing with the effects of cross-contamination; and (4) ensuring that the results are reproducible. We investigated these factors by generating sequence datasets, including biological and technical replicates, directly from clinical samples obtained from a small cohort of patients who had been infected congenitally with the herpesvirus human cytomegalovirus, with the aim of developing a strategy for identifying high-confidence intrahost variants. We found that such variants were few in number and typically present in low proportions and concluded that human cytomegalovirus exhibits a very low level of intrahost variability. In addition to clarifying the situation regarding human cytomegalovirus, our strategy has wider applicability to understanding the intrahost variability of other viruses.
Keywords: congenital infection; human cytomegalovirus; intrahost evolution; sequence variability.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
Similar articles
-
Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants.PLoS Pathog. 2011 May;7(5):e1001344. doi: 10.1371/journal.ppat.1001344. Epub 2011 May 19. PLoS Pathog. 2011. PMID: 21625576 Free PMC article.
-
Human Cytomegalovirus Genomes Sequenced Directly From Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss.J Infect Dis. 2019 Jul 31;220(5):781-791. doi: 10.1093/infdis/jiz208. J Infect Dis. 2019. PMID: 31050742 Free PMC article.
-
Deep-sequencing of viral genomes from a large and diverse cohort of treatment-naive HIV-infected persons shows associations between intrahost genetic diversity and viral load.PLoS Comput Biol. 2023 Jan 3;19(1):e1010756. doi: 10.1371/journal.pcbi.1010756. eCollection 2023 Jan. PLoS Comput Biol. 2023. PMID: 36595537 Free PMC article.
-
Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Cochrane Database Syst Rev. 2022 Feb 1;2(2022):CD014217. doi: 10.1002/14651858.CD014217. Cochrane Database Syst Rev. 2022. PMID: 36321557 Free PMC article.
-
Human cytomegalovirus intrahost evolution-a new avenue for understanding and controlling herpesvirus infections.Curr Opin Virol. 2014 Oct;8:109-15. doi: 10.1016/j.coviro.2014.08.001. Epub 2014 Aug 25. Curr Opin Virol. 2014. PMID: 25154343 Free PMC article. Review.
References
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
