

WaterKit: Thermodynamic Profiling of Protein Hydration Sites
- PMID: 37094087
- PMCID: PMC10732097
- DOI: 10.1021/acs.jctc.2c01087
WaterKit: Thermodynamic Profiling of Protein Hydration Sites
Abstract
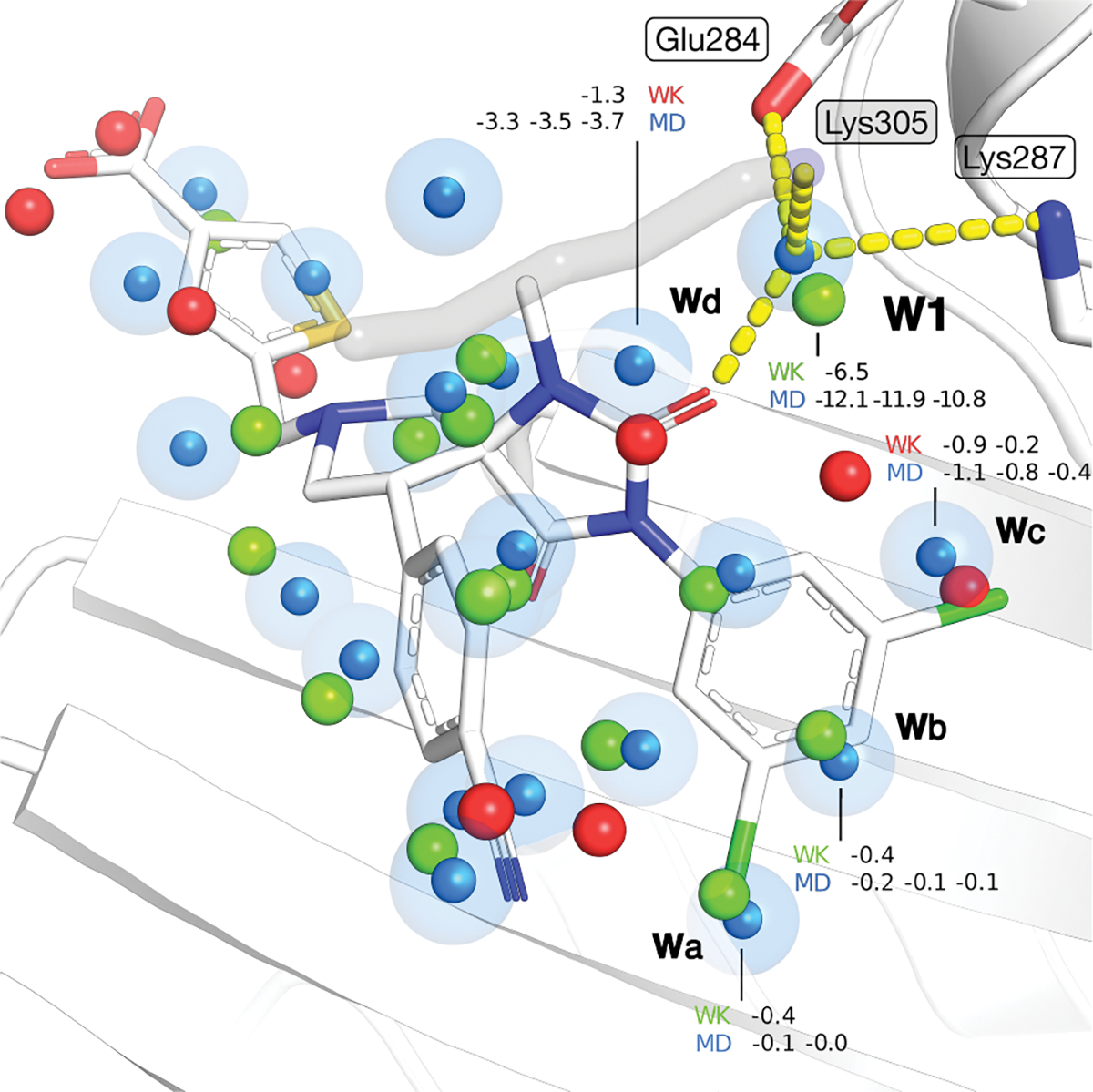
Water desolvation is one of the key components of the free energy of binding of small molecules to their receptors. Thus, understanding the energetic balance of solvation and desolvation resulting from individual water molecules can be crucial when estimating ligand binding, especially when evaluating different molecules and poses as done in High-Throughput Virtual Screening (HTVS). Over the most recent decades, several methods were developed to tackle this problem, ranging from fast approximate methods (usually empirical functions using either discrete atom-atom pairwise interactions or continuum solvent models) to more computationally expensive and accurate ones, mostly based on Molecular Dynamics (MD) simulations, such as Grid Inhomogeneous Solvation Theory (GIST) or Double Decoupling. On one hand, MD-based methods are prohibitive to use in HTVS to estimate the role of waters on the fly for each ligand. On the other hand, fast and approximate methods show an unsatisfactory level of accuracy, with low agreement with results obtained with the more expensive methods. Here we introduce WaterKit, a new grid-based sampling method with explicit water molecules to calculate thermodynamic properties using the GIST method. Our results show that the discrete placement of water molecules is successful in reproducing the position of crystallographic waters with very high accuracy, as well as providing thermodynamic estimates with accuracy comparable to more expensive MD simulations. Unlike these methods, WaterKit can be used to analyze specific regions on the protein surface, (such as the binding site of a receptor), without having to hydrate and simulate the whole receptor structure. The results show the feasibility of a general and fast method to compute thermodynamic properties of water molecules, making it well-suited to be integrated in high-throughput pipelines such as molecular docking.
Figures









Similar articles
-
AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking.Molecules. 2016 Nov 23;21(11):1604. doi: 10.3390/molecules21111604. Molecules. 2016. PMID: 27886114 Free PMC article.
-
Grid inhomogeneous solvation theory: hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril.J Chem Phys. 2012 Jul 28;137(4):044101. doi: 10.1063/1.4733951. J Chem Phys. 2012. PMID: 22852591 Free PMC article.
-
Calculation of Thermodynamic Properties of Bound Water Molecules.Methods Mol Biol. 2018;1762:389-402. doi: 10.1007/978-1-4939-7756-7_19. Methods Mol Biol. 2018. PMID: 29594782
-
Calculating Water Thermodynamics in the Binding Site of Proteins - Applications of WaterMap to Drug Discovery.Curr Top Med Chem. 2017;17(23):2586-2598. doi: 10.2174/1568026617666170414141452. Curr Top Med Chem. 2017. PMID: 28413953 Review.
-
Advances in the Treatment of Explicit Water Molecules in Docking and Binding Free Energy Calculations.Curr Med Chem. 2019;26(42):7598-7622. doi: 10.2174/0929867325666180514110824. Curr Med Chem. 2019. PMID: 29756561 Review.
Cited by
-
The structure and dynamics of water molecule networks underlie catalytic efficiency in a glycoside exo-hydrolase.Commun Biol. 2025 May 10;8(1):729. doi: 10.1038/s42003-025-08113-9. Commun Biol. 2025. PMID: 40348901 Free PMC article.
-
Integrating Quantum Mechanics into Protein-Ligand Docking: Toward Higher Accuracy and Reliability.Res Sq [Preprint]. 2024 Dec 5:rs.3.rs-5433993. doi: 10.21203/rs.3.rs-5433993/v1. Res Sq. 2024. PMID: 39678339 Free PMC article. Preprint.
-
An experimental proxy of water displaceability for ligand discovery.Nat Methods. 2025 Jul;22(7):1476-1485. doi: 10.1038/s41592-025-02724-0. Epub 2025 Jun 27. Nat Methods. 2025. PMID: 40579627
References
-
- Ben-Naim A, “Molecular recognition–viewed through the eyes of the solvent”, 101–102, pp. 309–319. - PubMed
-
- Spyrakis Francesca, Ahmed Mostafa H., Bayden Alexander S., Cozzini Pietro, Mozzarelli Andrea, and Kellogg Glen E., “The roles of water in the protein matrix: A largely untapped resource for drug discovery”, 60(16), pp. 6781–6827. - PubMed
-
- Dunitz Jack D., “The entropic cost of bound water in crystals and biomolecules”, 264(5159), pp. 670–670. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources