

Efficient engineering of human and mouse primary cells using peptide-assisted genome editing
- PMID: 37095348
- PMCID: PMC11230135
- DOI: 10.1038/s41587-023-01756-1
Efficient engineering of human and mouse primary cells using peptide-assisted genome editing
Abstract
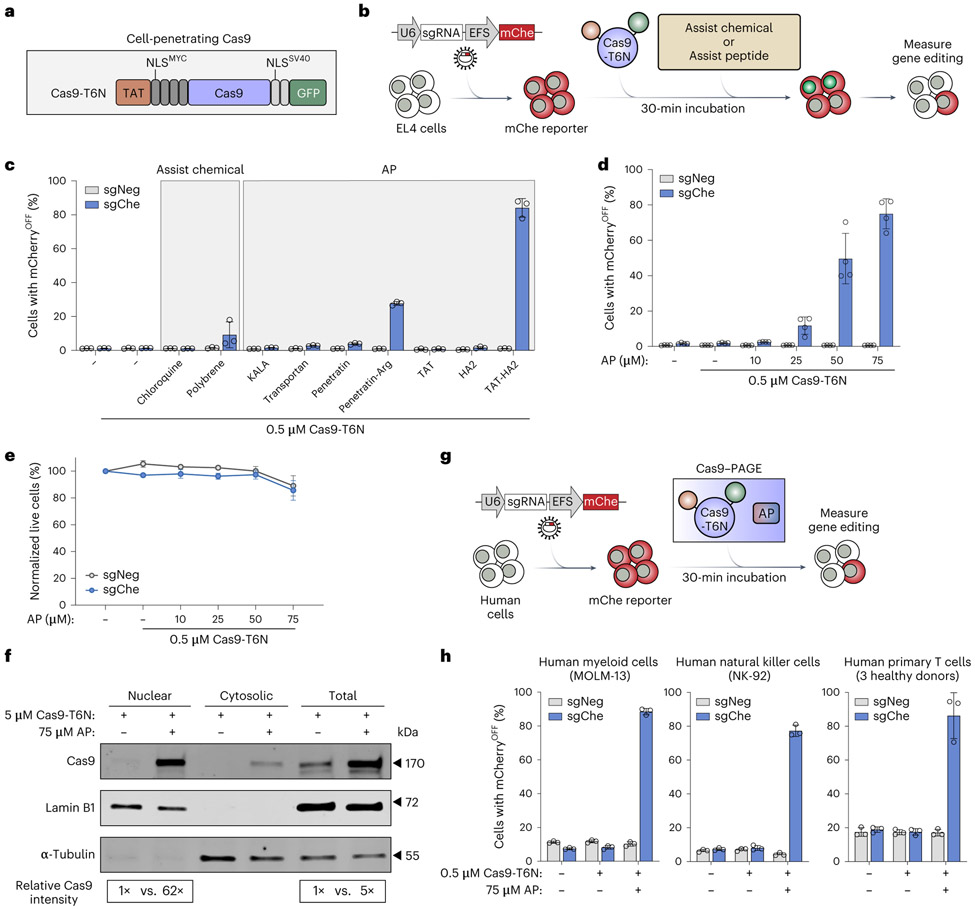
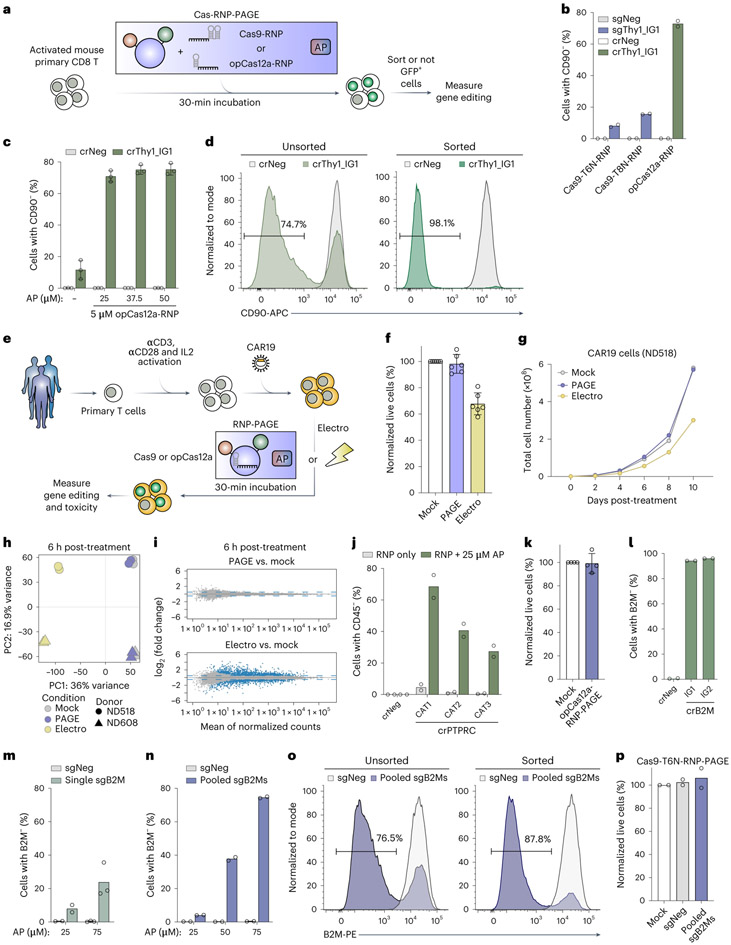
Simple, efficient and well-tolerated delivery of CRISPR genome editing systems into primary cells remains a major challenge. Here we describe an engineered Peptide-Assisted Genome Editing (PAGE) CRISPR-Cas system for rapid and robust editing of primary cells with minimal toxicity. The PAGE system requires only a 30-min incubation with a cell-penetrating Cas9 or Cas12a and a cell-penetrating endosomal escape peptide to achieve robust single and multiplex genome editing. Unlike electroporation-based methods, PAGE gene editing has low cellular toxicity and shows no significant transcriptional perturbation. We demonstrate rapid and efficient editing of primary cells, including human and mouse T cells, as well as human hematopoietic progenitor cells, with editing efficiencies upwards of 98%. PAGE provides a broadly generalizable platform for next-generation genome engineering in primary cells.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures


Comment in
-
Delivering genome editing tools to primary cells.Nat Rev Drug Discov. 2023 Jun;22(6):444. doi: 10.1038/d41573-023-00069-x. Nat Rev Drug Discov. 2023. PMID: 37142697 No abstract available.
References
-
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S & Milone MC CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
