

Efficient evolution of human antibodies from general protein language models
- PMID: 37095349
- PMCID: PMC10869273
- DOI: 10.1038/s41587-023-01763-2
Efficient evolution of human antibodies from general protein language models
Abstract
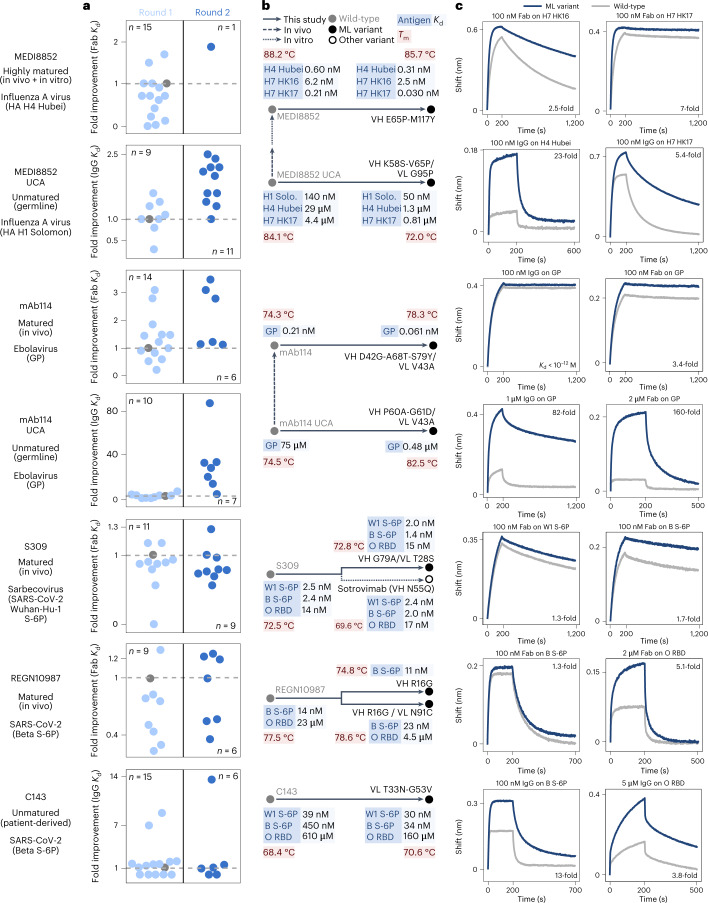
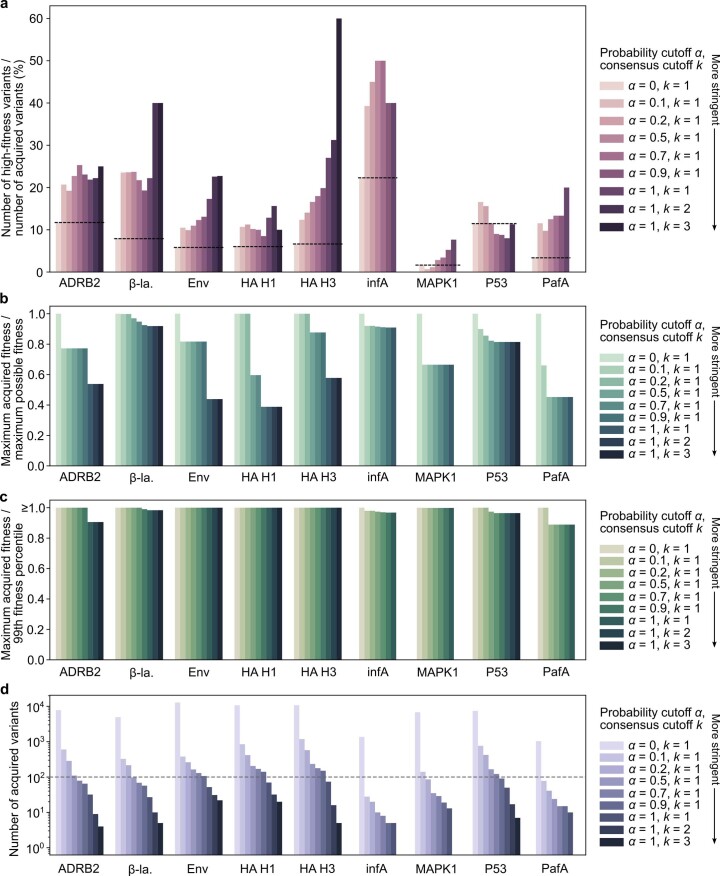
Natural evolution must explore a vast landscape of possible sequences for desirable yet rare mutations, suggesting that learning from natural evolutionary strategies could guide artificial evolution. Here we report that general protein language models can efficiently evolve human antibodies by suggesting mutations that are evolutionarily plausible, despite providing the model with no information about the target antigen, binding specificity or protein structure. We performed language-model-guided affinity maturation of seven antibodies, screening 20 or fewer variants of each antibody across only two rounds of laboratory evolution, and improved the binding affinities of four clinically relevant, highly mature antibodies up to sevenfold and three unmatured antibodies up to 160-fold, with many designs also demonstrating favorable thermostability and viral neutralization activity against Ebola and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pseudoviruses. The same models that improve antibody binding also guide efficient evolution across diverse protein families and selection pressures, including antibiotic resistance and enzyme activity, suggesting that these results generalize to many settings.
© 2023. The Author(s).
Conflict of interest statement
B.L.H., V.R.S. and P.S.K. are named as inventors on a provisional patent application applied for by Stanford University and the Chan Zuckerberg Biohub related to this study. B.L.H. performs research for Meta Platforms, Inc. The remaining authors declare no competing interests.
Figures









References
-
- Futuyma, D. J. Evolutionary Biology 3rd ed (Sinauer Associates, 1997).
-
- Wright, S. The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc. of the VI International Congress of Genetics 355–366 (Blackwell, 1932).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
