

Beginner's Guide on the Use of PAML to Detect Positive Selection
- PMID: 37096789
- PMCID: PMC10127084
- DOI: 10.1093/molbev/msad041
Beginner's Guide on the Use of PAML to Detect Positive Selection
Abstract
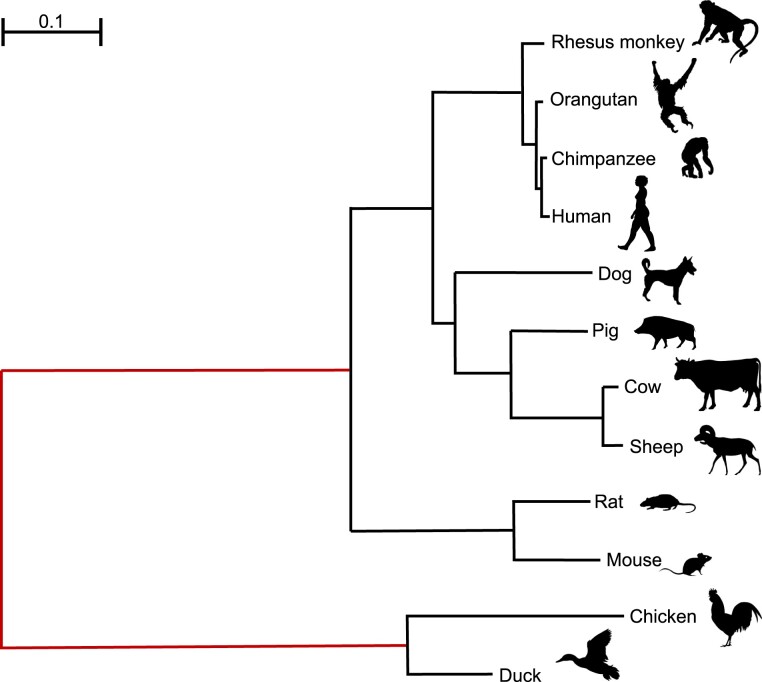
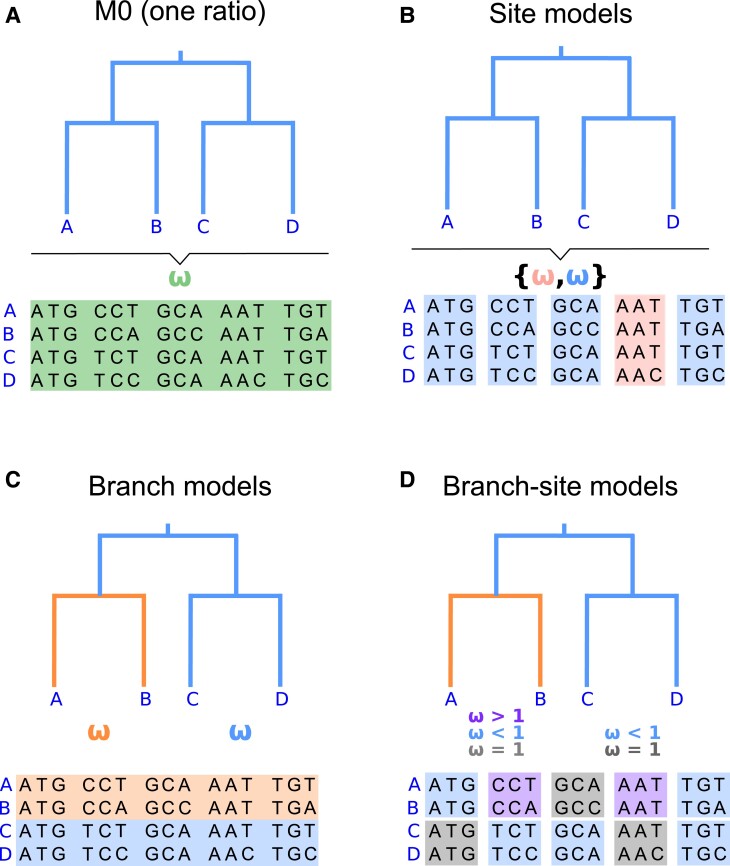
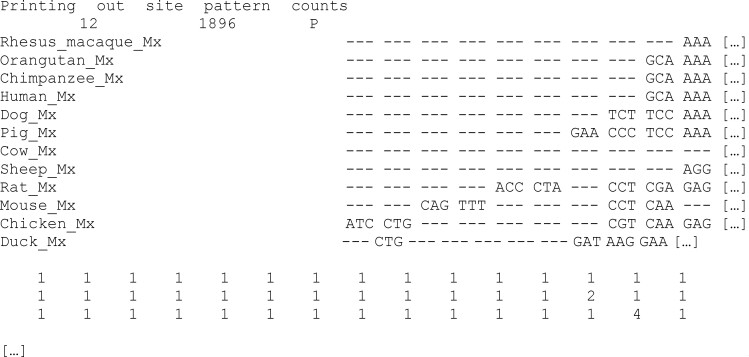
The CODEML program in the PAML package has been widely used to analyze protein-coding gene sequences to estimate the synonymous and nonsynonymous rates (dS and dN) and to detect positive Darwinian selection driving protein evolution. For users not familiar with molecular evolutionary analysis, the program is known to have a steep learning curve. Here, we provide a step-by-step protocol to illustrate the commonly used tests available in the program, including the branch models, the site models, and the branch-site models, which can be used to detect positive selection driving adaptive protein evolution affecting particular lineages of the species phylogeny, affecting a subset of amino acid residues in the protein, and affecting a subset of sites along prespecified lineages, respectively. A data set of the myxovirus (Mx) genes from ten mammal and two bird species is used as an example. We discuss a new feature in CODEML that allows users to perform positive selection tests for multiple genes for the same set of taxa, as is common in modern genome-sequencing projects. The PAML package is distributed at https://github.com/abacus-gene/paml under the GNU license, with support provided at its discussion site (https://groups.google.com/g/pamlsoftware). Data files used in this protocol are available at https://github.com/abacus-gene/paml-tutorial.
Keywords: d N/dS; PAML; adaptive evolution; nonsynonymous substitutions; positive selection; synonymous substitutions.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures





References
-
- Anisimova M, Kosiol C. 2009. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 26:255–271. - PubMed
-
- Anisimova M, Yang Z. 2007. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 24:1219–1228. - PubMed
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 57:289–300.
-
- Benjamini Y, Hochberg Y. 2000. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educat Behav Stat. 25:83.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
