

NSD1 deposits histone H3 lysine 36 dimethylation to pattern non-CG DNA methylation in neurons
- PMID: 37098340
- PMCID: PMC10230755
- DOI: 10.1016/j.molcel.2023.04.001
NSD1 deposits histone H3 lysine 36 dimethylation to pattern non-CG DNA methylation in neurons
Abstract
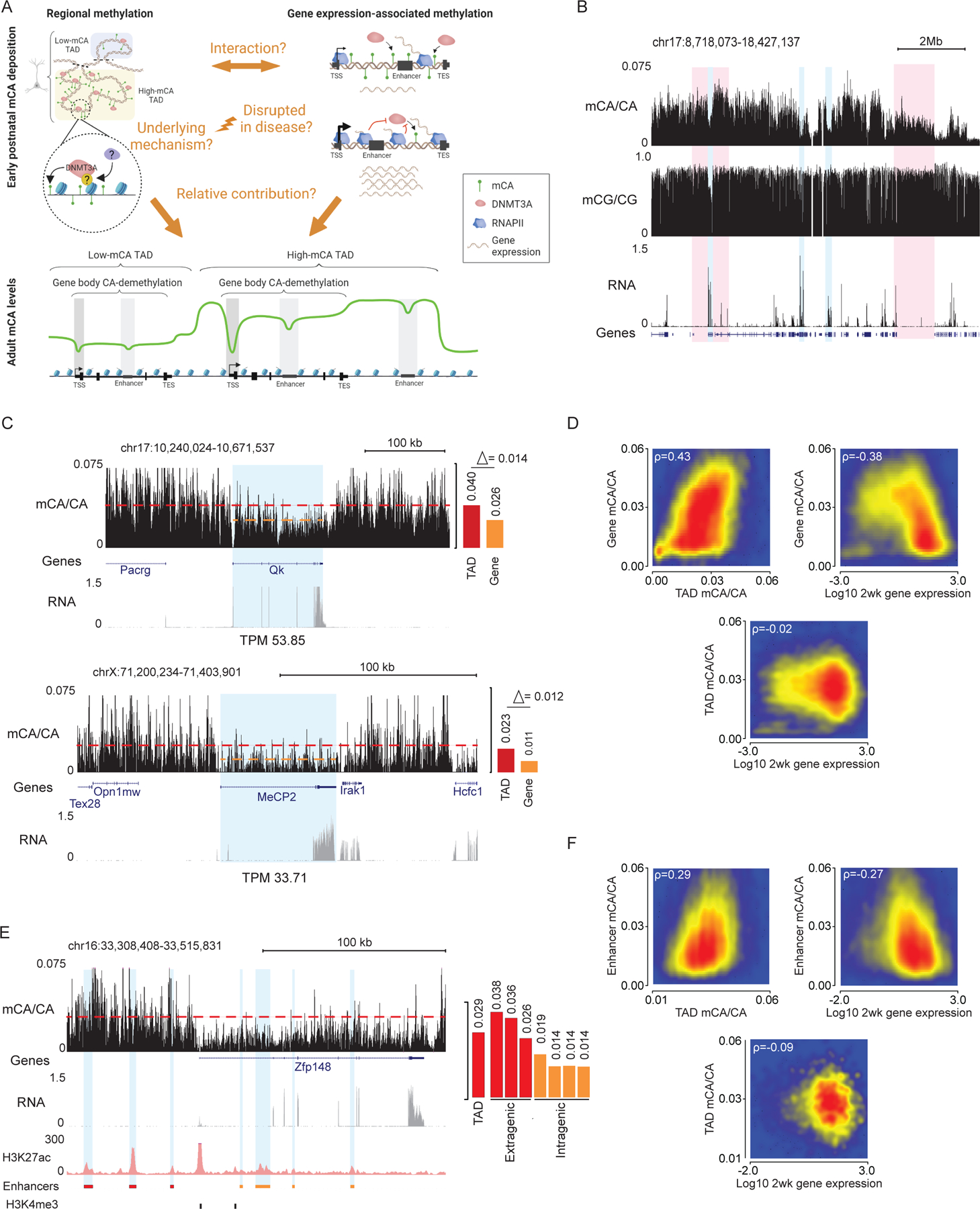
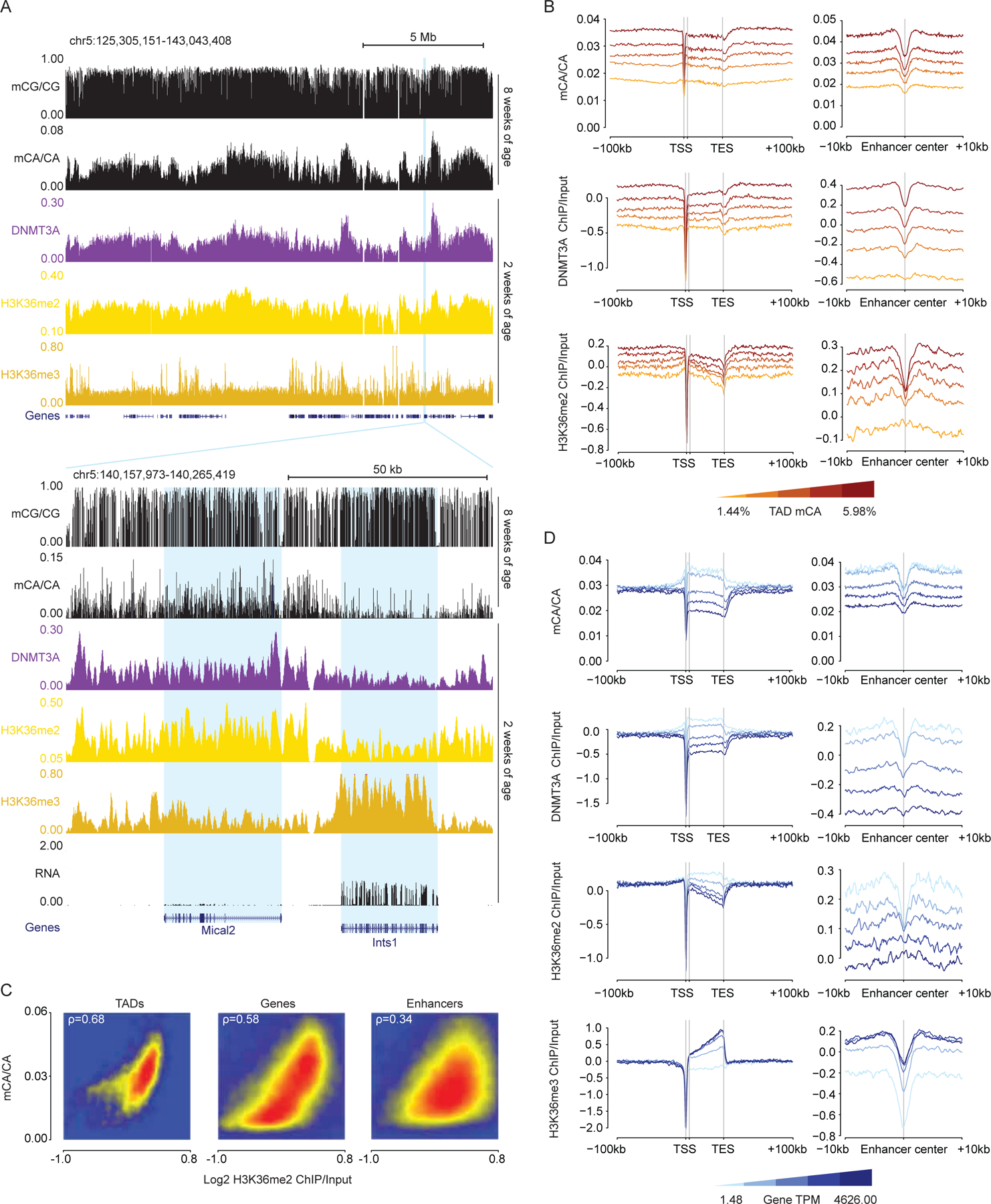
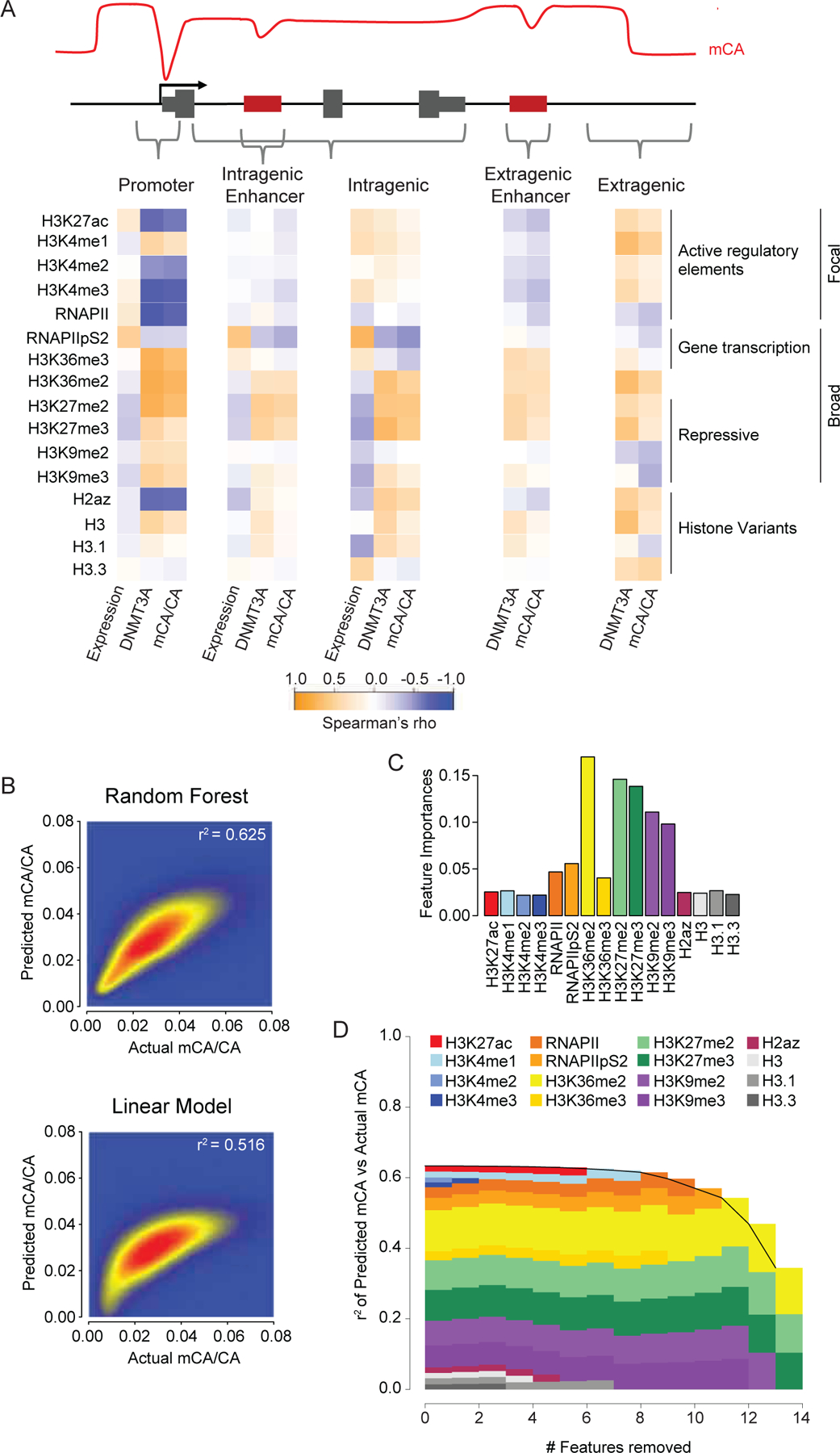
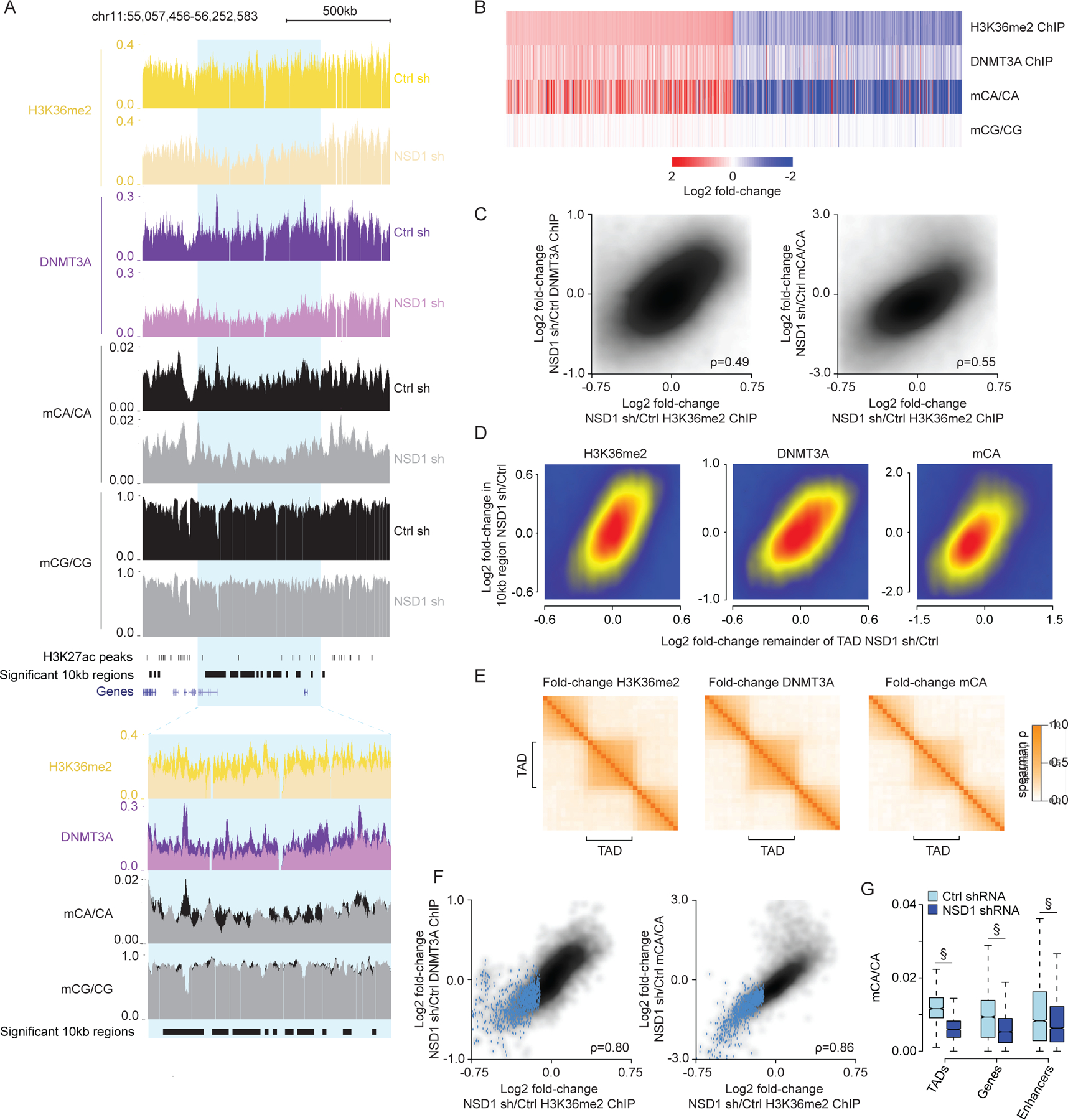
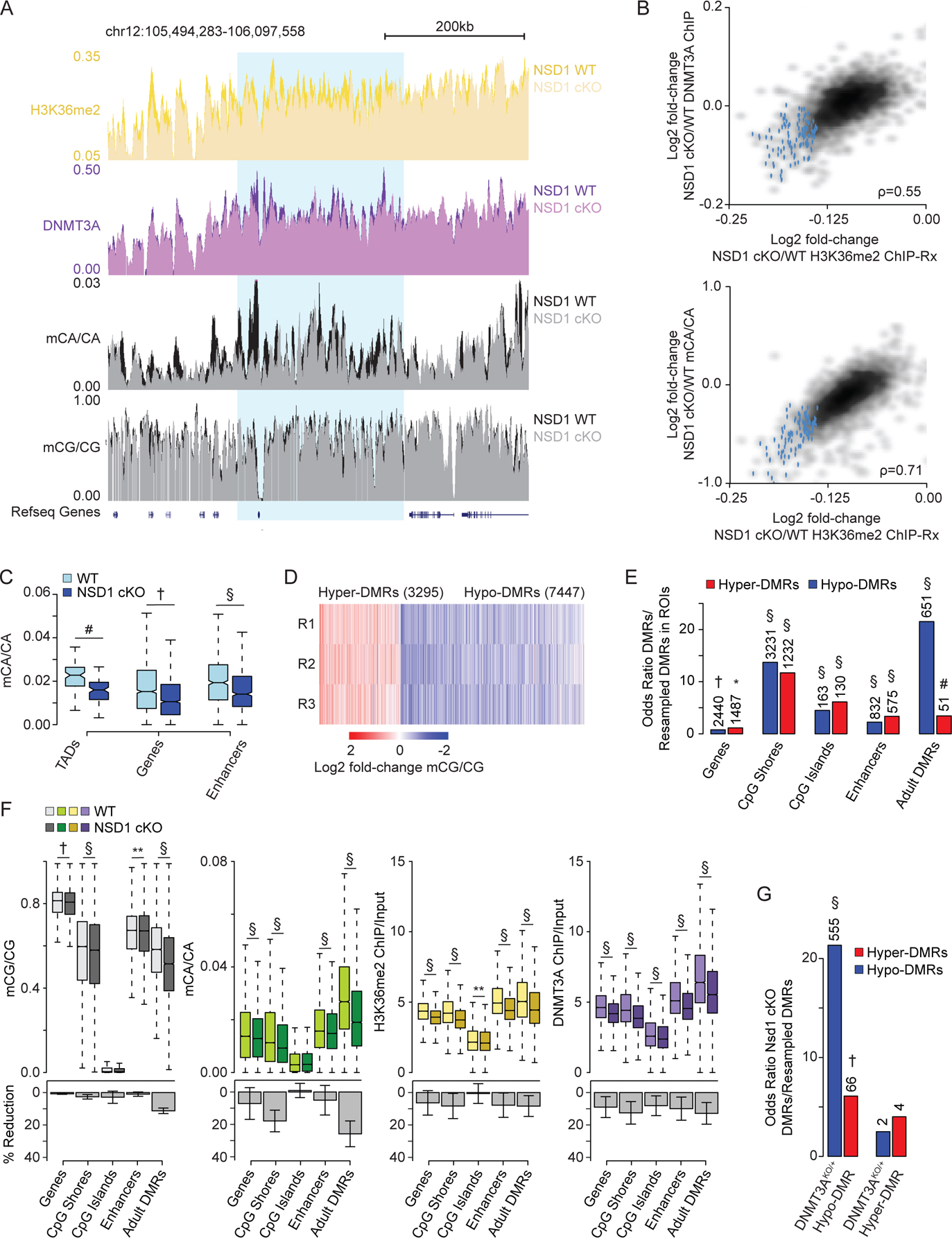
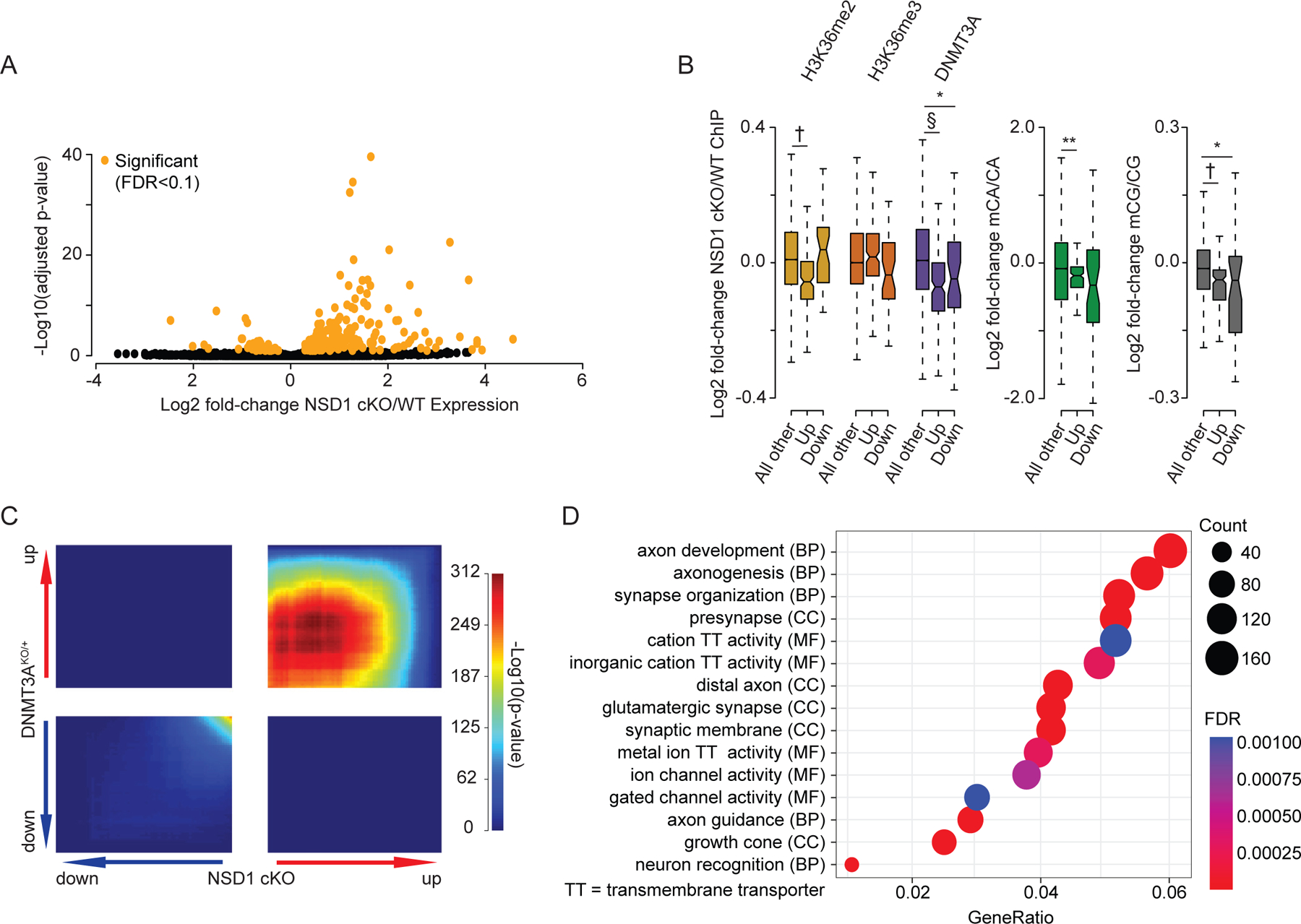
During postnatal development, the DNA methyltransferase DNMT3A deposits high levels of non-CG cytosine methylation in neurons. This methylation is critical for transcriptional regulation, and loss of this mark is implicated in DNMT3A-associated neurodevelopmental disorders (NDDs). Here, we show in mice that genome topology and gene expression converge to shape histone H3 lysine 36 dimethylation (H3K36me2) profiles, which in turn recruit DNMT3A and pattern neuronal non-CG methylation. We show that NSD1, an H3K36 methyltransferase mutated in NDD, is required for the patterning of megabase-scale H3K36me2 and non-CG methylation in neurons. We find that brain-specific deletion of NSD1 causes altered DNA methylation that overlaps with DNMT3A disorder models to drive convergent dysregulation of key neuronal genes that may underlie shared phenotypes in NSD1- and DNMT3A-associated NDDs. Our findings indicate that H3K36me2 deposited by NSD1 is important for neuronal non-CG DNA methylation and suggest that the H3K36me2-DNMT3A-non-CG-methylation pathway is likely disrupted in NSD1-associated NDDs.
Keywords: DNA methylation; DNMT3A; NSD1; Sotos syndrome; genome topology; histone methylation; neurodevelopmental disease; non-CG methylation.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
NSD1 deposits histone H3 lysine 36 dimethylation to pattern non-CG DNA methylation in neurons.bioRxiv [Preprint]. 2023 Feb 17:2023.02.17.528965. doi: 10.1101/2023.02.17.528965. bioRxiv. 2023. Update in: Mol Cell. 2023 May 4;83(9):1412-1428.e7. doi: 10.1016/j.molcel.2023.04.001. PMID: 36824816 Free PMC article. Updated. Preprint.
References
-
- Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, Yu M, He C, Sanguinetti G, Sowers LC, et al. (2017). MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. Plos Genet 13, e1006793. 10.1371/journal.pgen.1006793. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
