

Olipudase alfa enzyme replacement therapy for acid sphingomyelinase deficiency (ASMD): sustained improvements in clinical outcomes after 6.5 years of treatment in adults
- PMID: 37098529
- PMCID: PMC10131350
- DOI: 10.1186/s13023-023-02700-x
Olipudase alfa enzyme replacement therapy for acid sphingomyelinase deficiency (ASMD): sustained improvements in clinical outcomes after 6.5 years of treatment in adults
Abstract
Background: Enzyme replacement therapy with olipudase alfa, a recombinant human acid sphingomyelinase (rhASM), is indicated for non-central nervous system manifestations of acid sphingomyelinase deficiency (ASMD) in children and adults. An ongoing, open-label, long-term study (NCT02004704) assessed the safety and efficacy of olipudase alfa in 5 adults with ASMD.
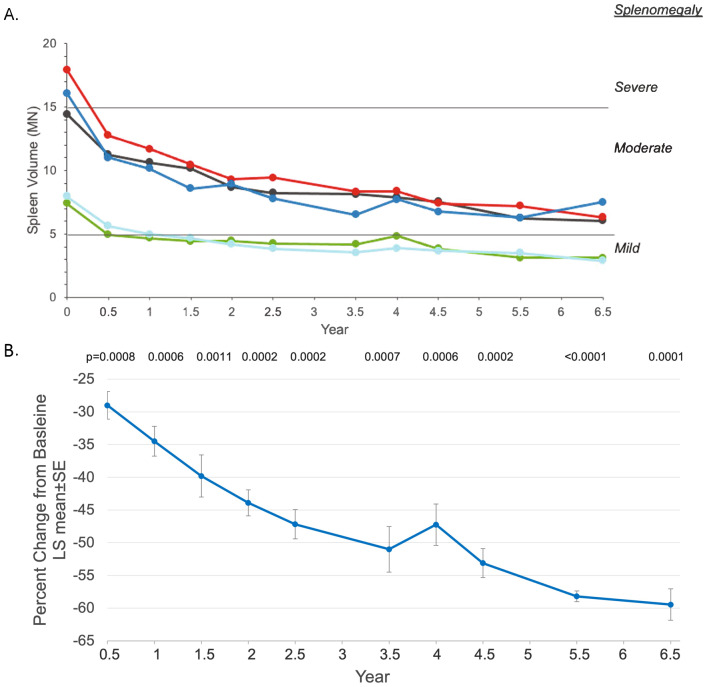
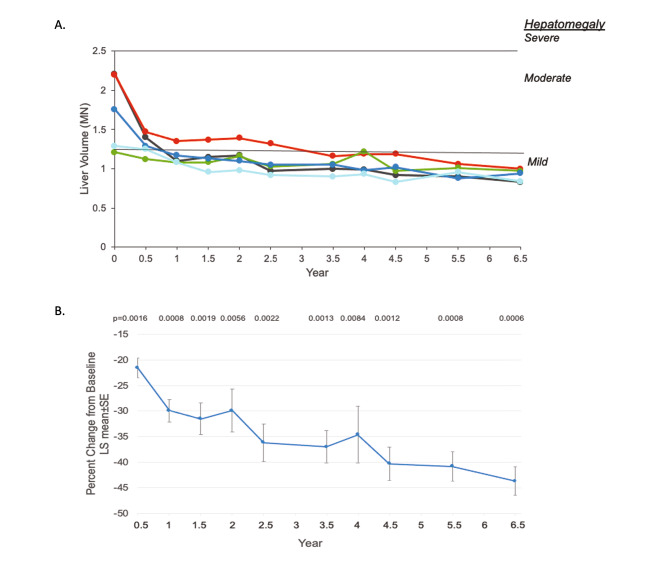
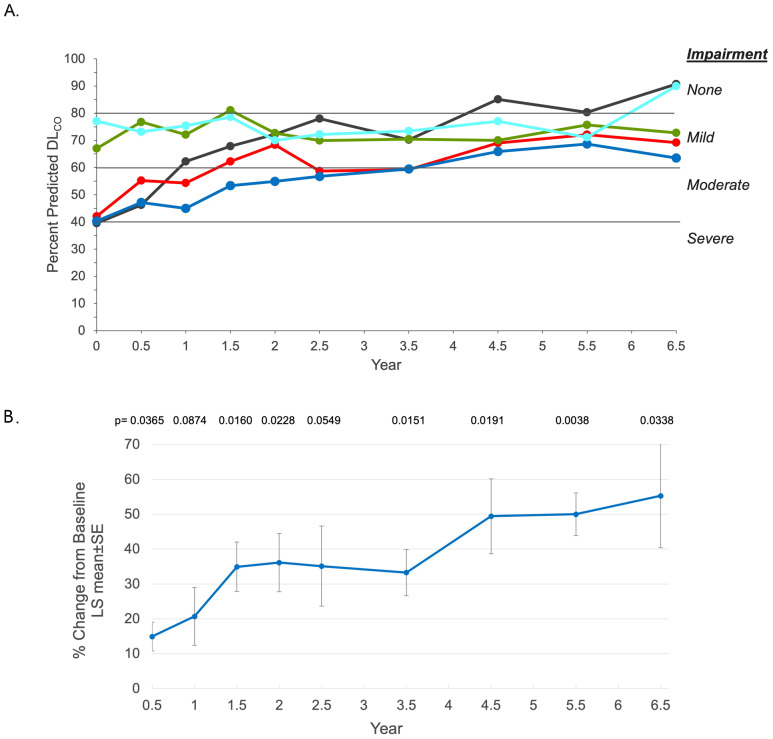
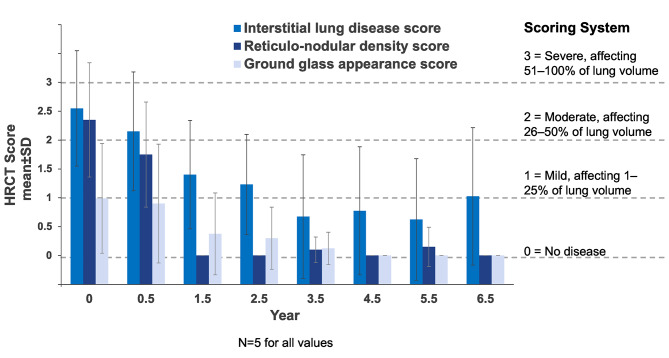
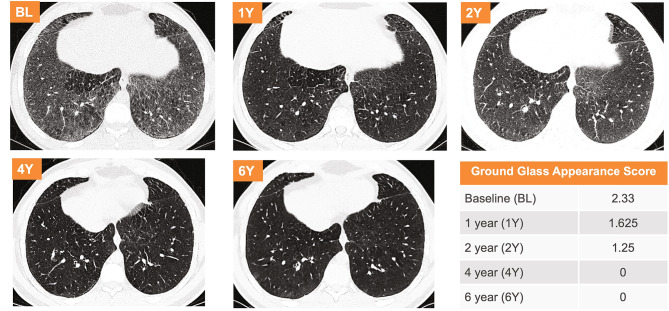
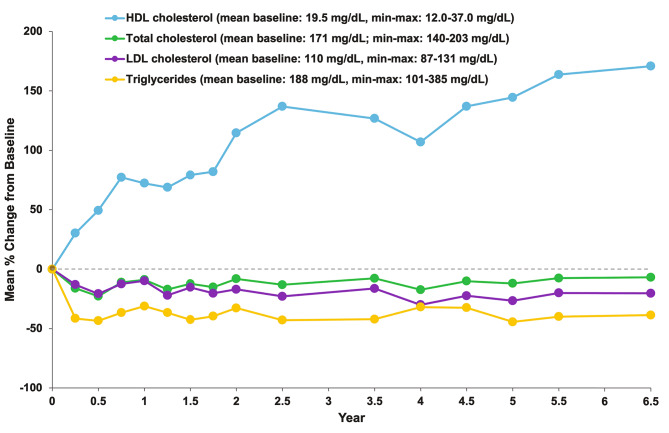
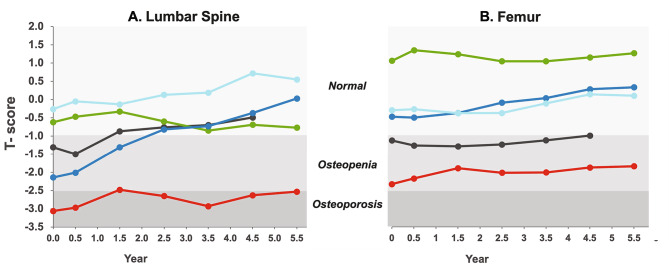
Results: After 6.5 years of treatment, there were no discontinuations, no olipudase-alfa-related serious adverse events, and no new safety signals compared to earlier assessments. Most treatment-emergent adverse events were mild in intensity (1742/1766, 98.6%). Among treatment-related adverse events (n = 657), more than half were considered infusion-associated reactions (n = 403, 61.3%) such as headache, nausea, abdominal pain, arthralgia, pyrexia, and fatigue. No patient developed neutralizing anti-drug antibodies to cellular uptake, and there were no clinically significant adverse changes in vital signs, hematology, or cardiac safety parameters. Improvements (decreases) in spleen and liver volumes progressed through 6.5 years (mean changes from baseline of -59.5% and -43.7%, respectively). There was a mean increase in diffusing capacity of the lung for carbon monoxide from baseline of 55.3%, accompanied by improvements in interstitial lung disease parameters. Lipid profiles at baseline indicated dyslipidemia. All patients had sustained decreases in pro-atherogenic lipid levels and increases in anti-atherogenic lipid levels following olipudase alfa treatment.
Conclusions: Olipudase alfa is the first disease-specific treatment for ASMD. This study demonstrates that long-term treatment with olipudase alfa is well-tolerated and is associated with sustained improvements in relevant disease clinical measures. NCT02004704 registered 26 November 2013, https://clinicaltrials.gov/ct2/show/NCT02004704?term=NCT02004704&draw=2&rank=1 .
Keywords: Acid sphingomyelinase deficiency; Niemann-Pick disease type B and type A/B; Olipudase alfa; Recombinant human acid sphingomyelinase.
© 2023. The Author(s).
Conflict of interest statement
RHL, MPW, and GAD have received consulting fees, honoraria and travel support to attend scientific meetings from Sanofi.
NA, AY, YK, and MK are employees of Sanofi and may own Sanofi stock.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
