

Sequence-structure-function relationships in the microbial protein universe
- PMID: 37100781
- PMCID: PMC10133388
- DOI: 10.1038/s41467-023-37896-w
Sequence-structure-function relationships in the microbial protein universe
Abstract
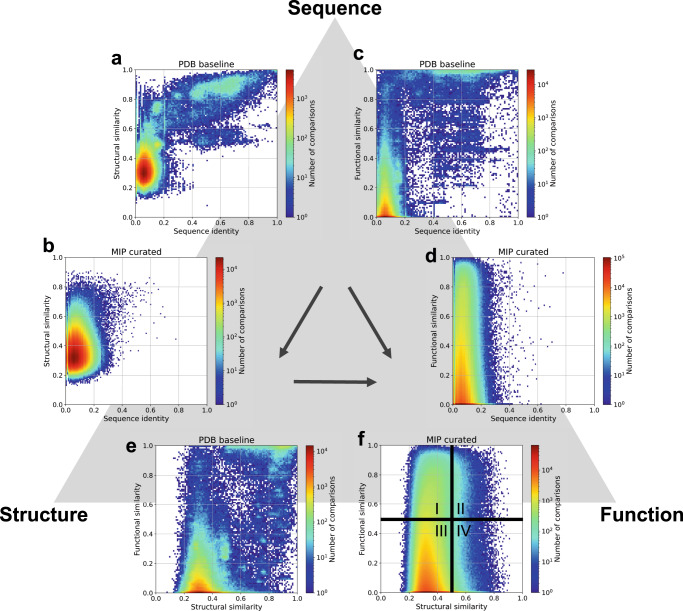
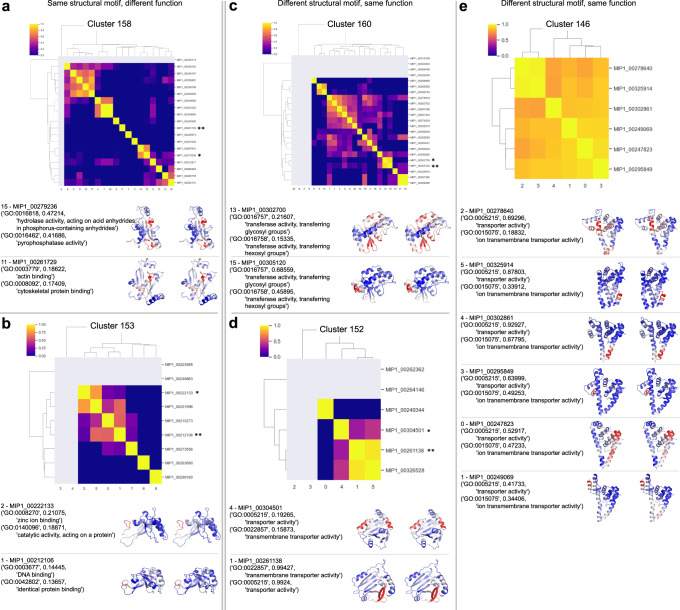
For the past half-century, structural biologists relied on the notion that similar protein sequences give rise to similar structures and functions. While this assumption has driven research to explore certain parts of the protein universe, it disregards spaces that don't rely on this assumption. Here we explore areas of the protein universe where similar protein functions can be achieved by different sequences and different structures. We predict ~200,000 structures for diverse protein sequences from 1,003 representative genomes across the microbial tree of life and annotate them functionally on a per-residue basis. Structure prediction is accomplished using the World Community Grid, a large-scale citizen science initiative. The resulting database of structural models is complementary to the AlphaFold database, with regards to domains of life as well as sequence diversity and sequence length. We identify 148 novel folds and describe examples where we map specific functions to structural motifs. We also show that the structural space is continuous and largely saturated, highlighting the need for a shift in focus across all branches of biology, from obtaining structures to putting them into context and from sequence-based to sequence-structure-function based meta-omics analyses.
© 2023. The Author(s).
Conflict of interest statement
R.B., V.G., and D.B. are currently working at Genentech and no explicit conflicts of interest result from this change in affiliation. All other authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
