

Integrating gene annotation with orthology inference at scale
- PMID: 37104600
- PMCID: PMC10193443
- DOI: 10.1126/science.abn3107
Integrating gene annotation with orthology inference at scale
Abstract
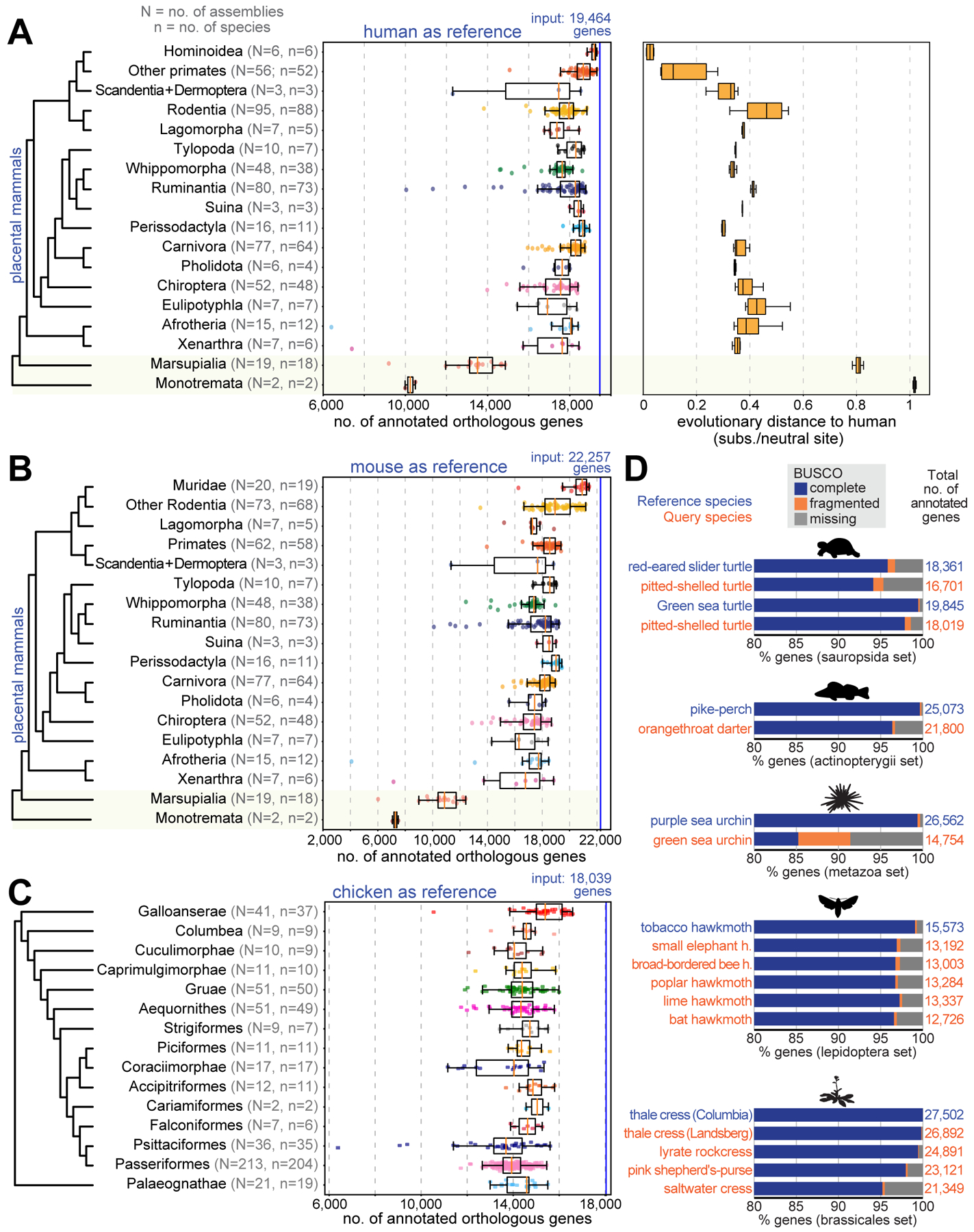
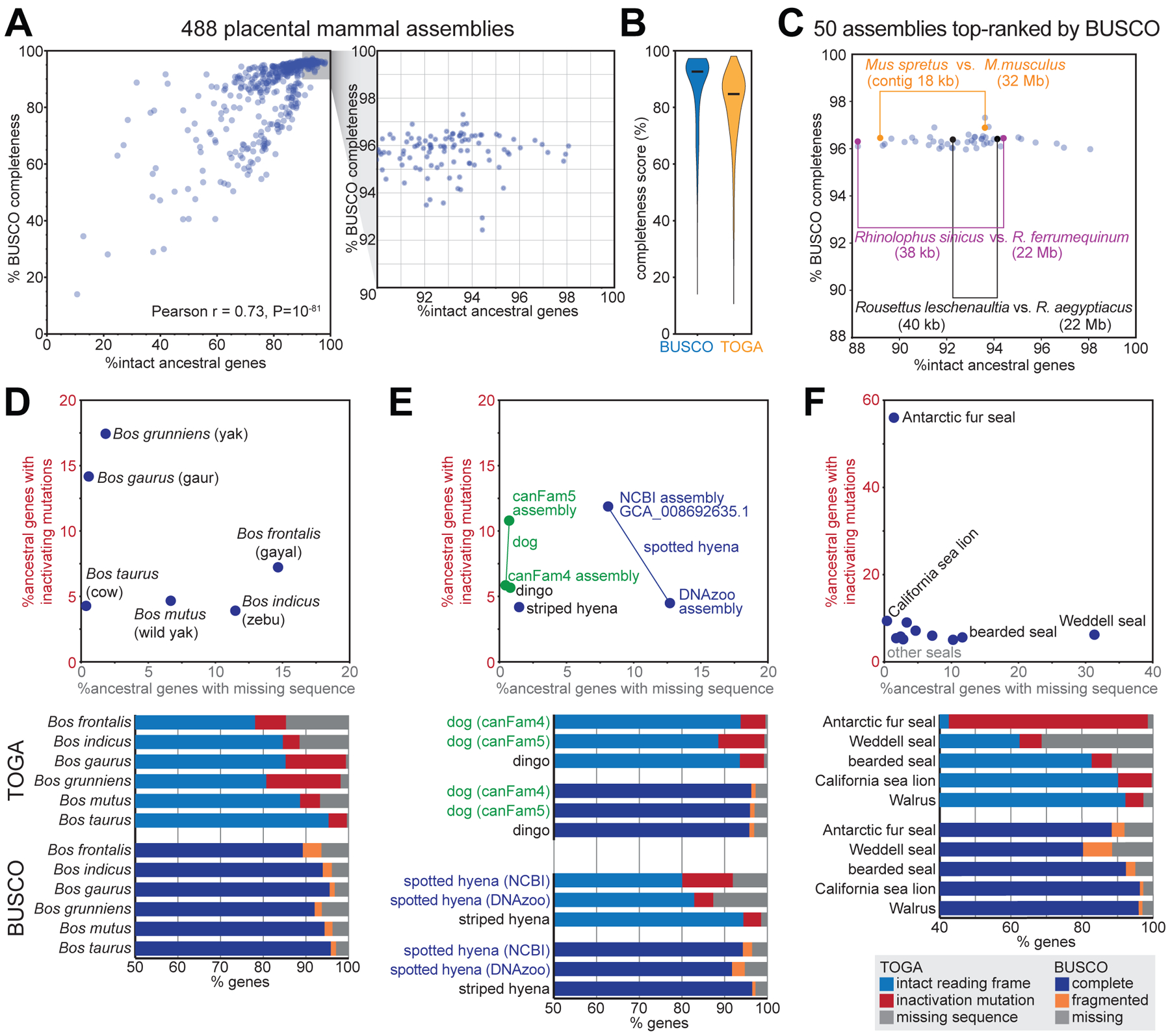
Annotating coding genes and inferring orthologs are two classical challenges in genomics and evolutionary biology that have traditionally been approached separately, limiting scalability. We present TOGA (Tool to infer Orthologs from Genome Alignments), a method that integrates structural gene annotation and orthology inference. TOGA implements a different paradigm to infer orthologous loci, improves ortholog detection and annotation of conserved genes compared with state-of-the-art methods, and handles even highly fragmented assemblies. TOGA scales to hundreds of genomes, which we demonstrate by applying it to 488 placental mammal and 501 bird assemblies, creating the largest comparative gene resources so far. Additionally, TOGA detects gene losses, enables selection screens, and automatically provides a superior measure of mammalian genome quality. TOGA is a powerful and scalable method to annotate and compare genes in the genomic era.
Conflict of interest statement
Figures
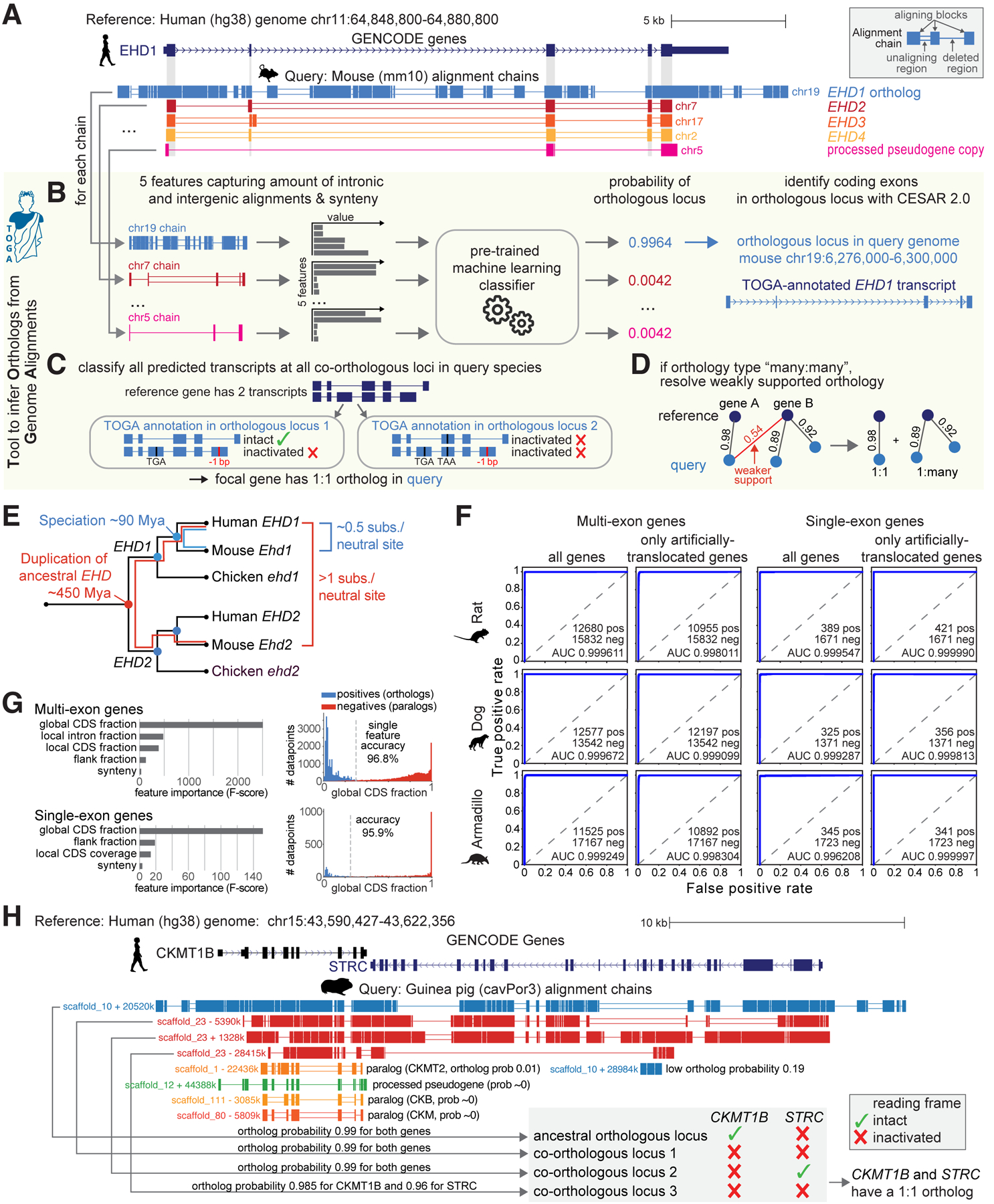
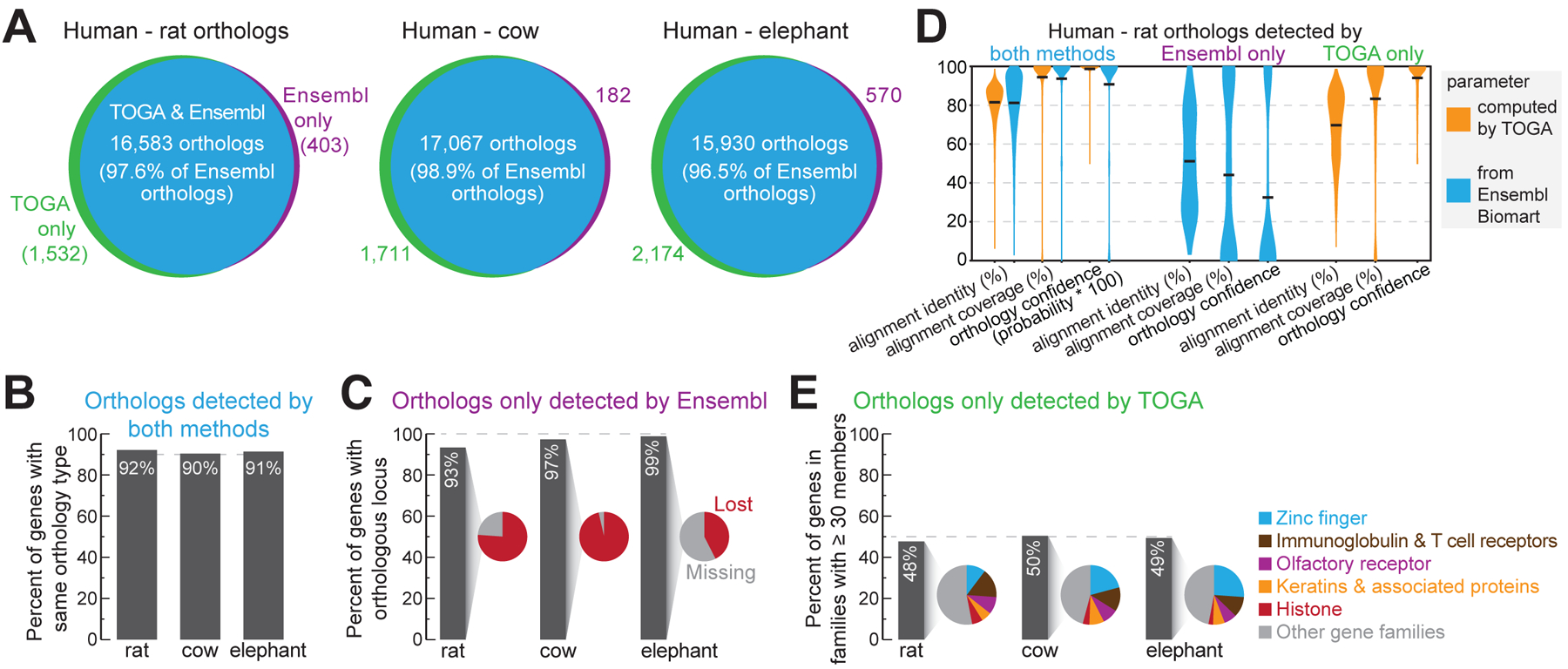
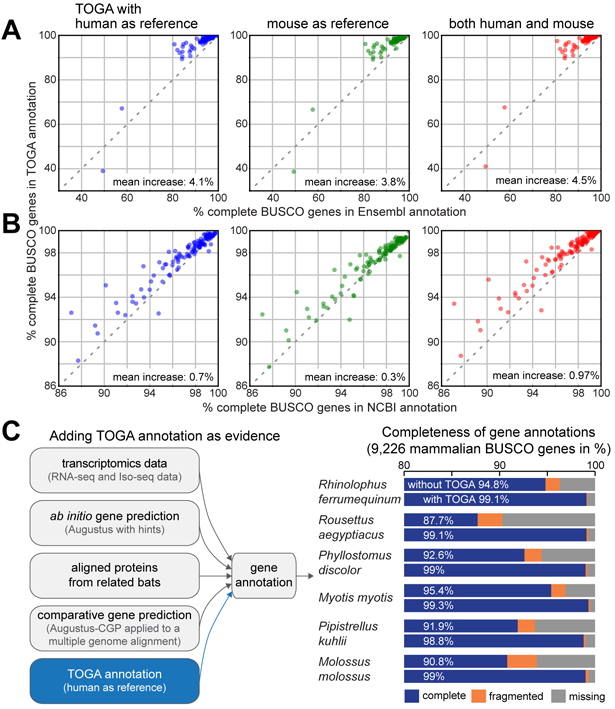
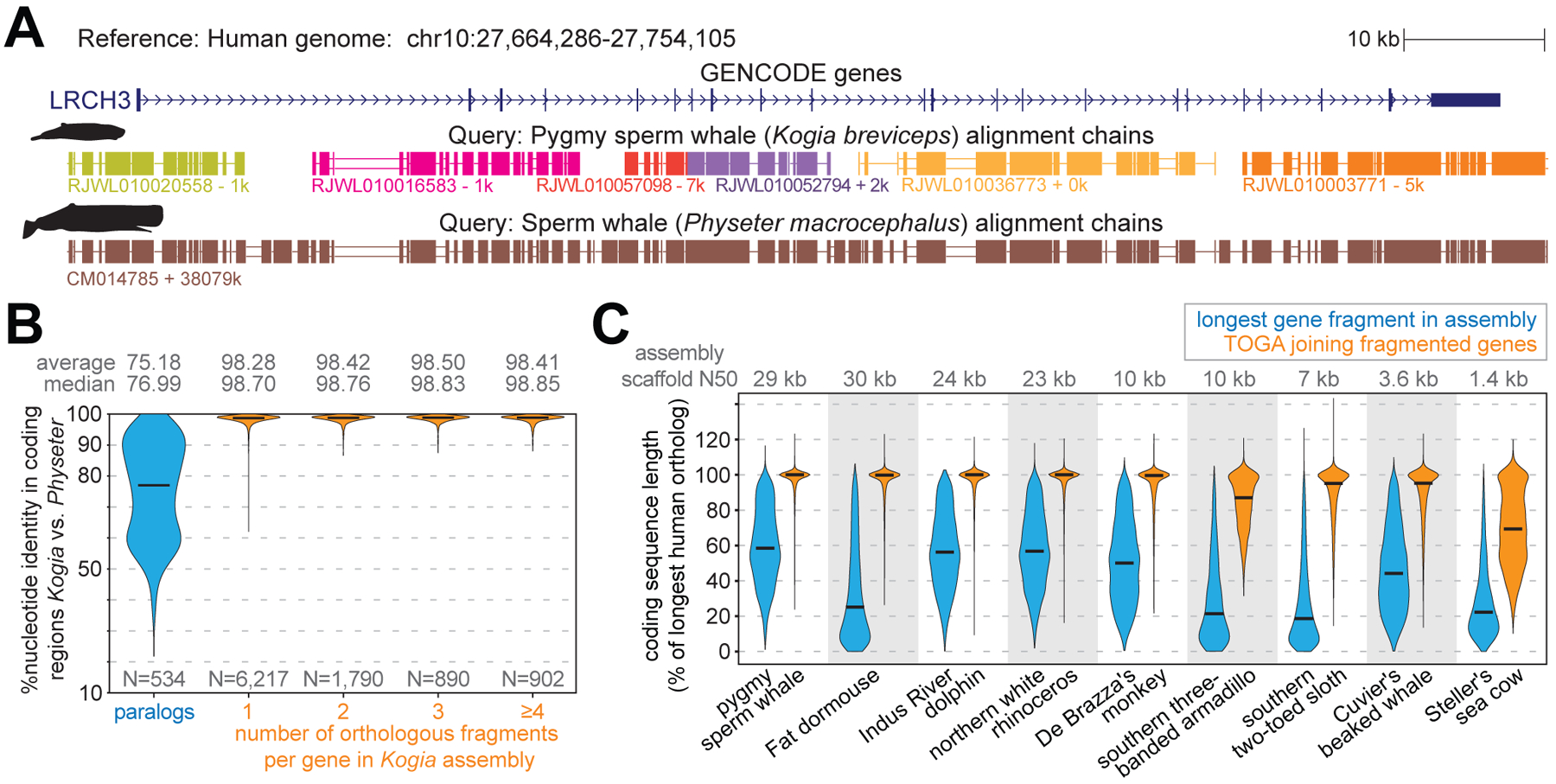
References
-
- Kapli P, Yang Z, Telford MJ, Phylogenetic tree building in the genomic age. Nature reviews. Genetics 21, 428–444 (2020). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
