

CNV-ClinViewer: enhancing the clinical interpretation of large copy-number variants online
- PMID: 37104749
- PMCID: PMC10174702
- DOI: 10.1093/bioinformatics/btad290
CNV-ClinViewer: enhancing the clinical interpretation of large copy-number variants online
Abstract
Motivation: Pathogenic copy-number variants (CNVs) can cause a heterogeneous spectrum of rare and severe disorders. However, most CNVs are benign and are part of natural variation in human genomes. CNV pathogenicity classification, genotype-phenotype analyses, and therapeutic target identification are challenging and time-consuming tasks that require the integration and analysis of information from multiple scattered sources by experts.
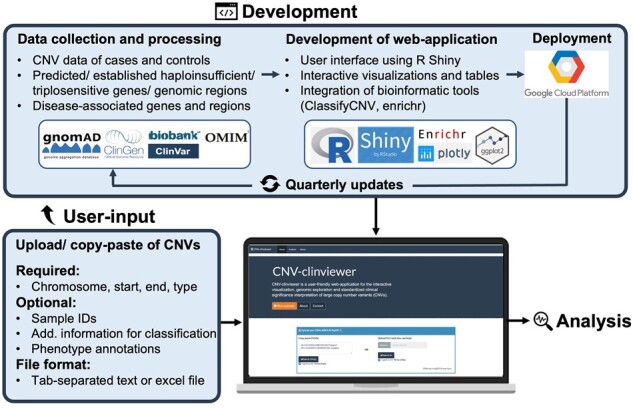
Results: Here, we introduce the CNV-ClinViewer, an open-source web application for clinical evaluation and visual exploration of CNVs. The application enables real-time interactive exploration of large CNV datasets in a user-friendly designed interface and facilitates semi-automated clinical CNV interpretation following the ACMG guidelines by integrating the ClassifCNV tool. In combination with clinical judgment, the application enables clinicians and researchers to formulate novel hypotheses and guide their decision-making process. Subsequently, the CNV-ClinViewer enhances for clinical investigators' patient care and for basic scientists' translational genomic research.
Availability and implementation: The web application is freely available at https://cnv-ClinViewer.broadinstitute.org and the open-source code can be found at https://github.com/LalResearchGroup/CNV-clinviewer.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures



References
-
- Babione JN, Ocampo W, Haubrich S. et al. Human-centred design processes for clinical decision support: a pulmonary embolism case study. Int J Med Inform 2020;142:104196. - PubMed
-
- Cai CJ et al. 2019. Human-centered tools for coping with imperfect algorithms during medical decision-making. In: Proceedings of the 2019 CHI Conference on Human Factors in Computing Systems, CHI ’19, pp. 1–14. Association for Computing Machinery, New York, NY, USA.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
