

Identification and Functional Analysis of Known and New Mutations in the Transcription Factor KLF1 Linked with β-Thalassemia-like Phenotypes
- PMID: 37106711
- PMCID: PMC10135830
- DOI: 10.3390/biology12040510
Identification and Functional Analysis of Known and New Mutations in the Transcription Factor KLF1 Linked with β-Thalassemia-like Phenotypes
Abstract
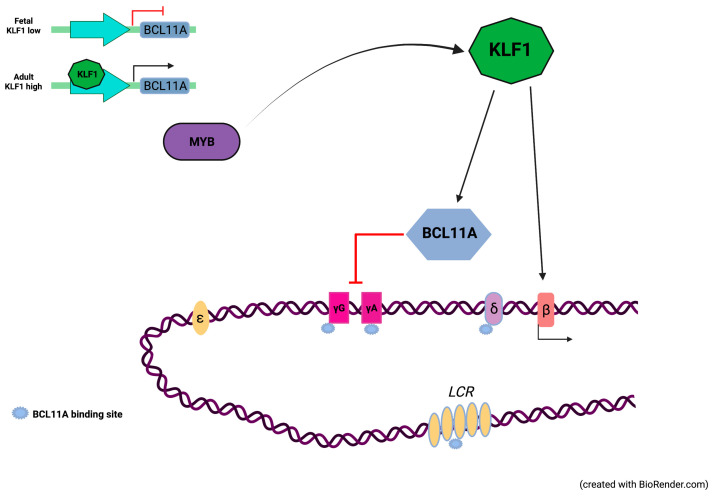
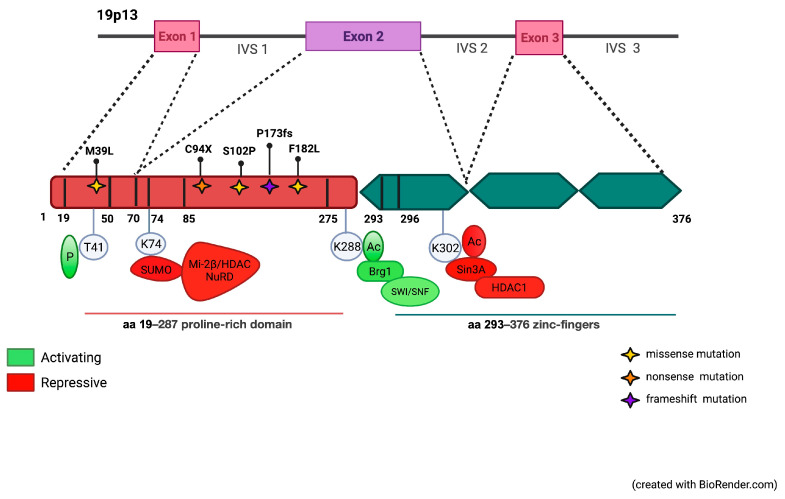
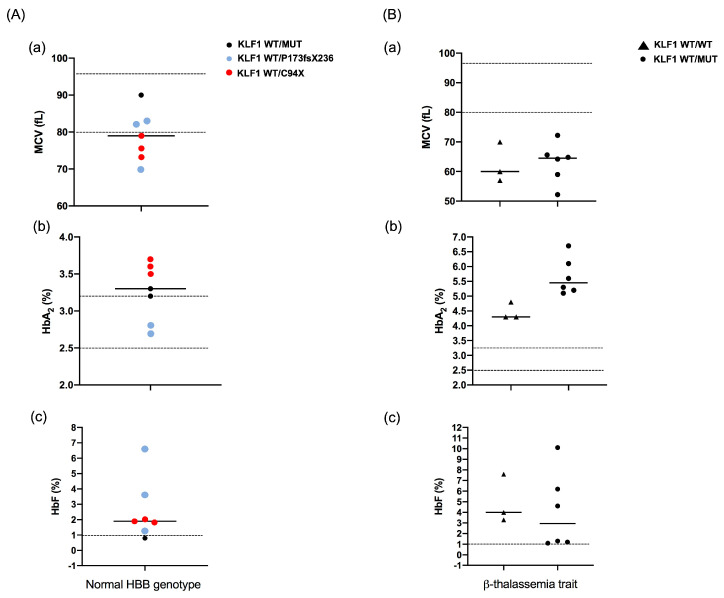
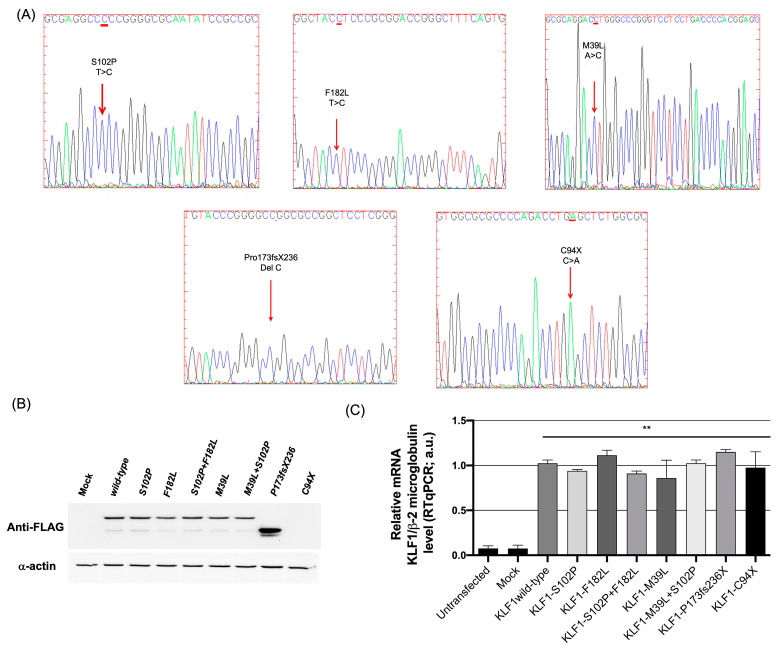
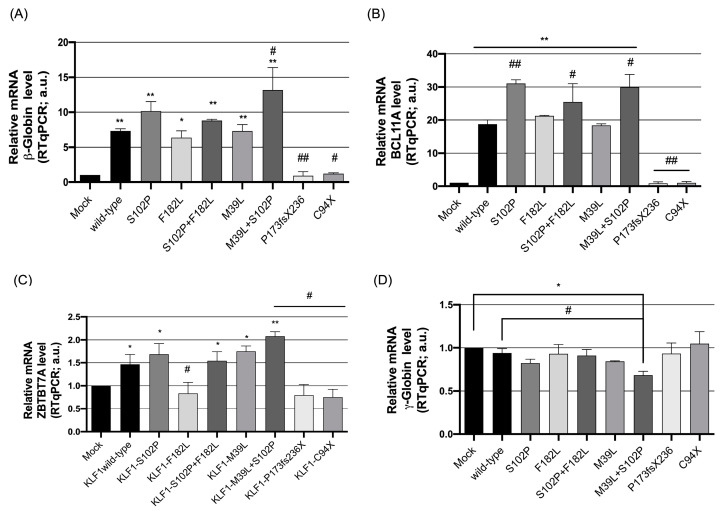
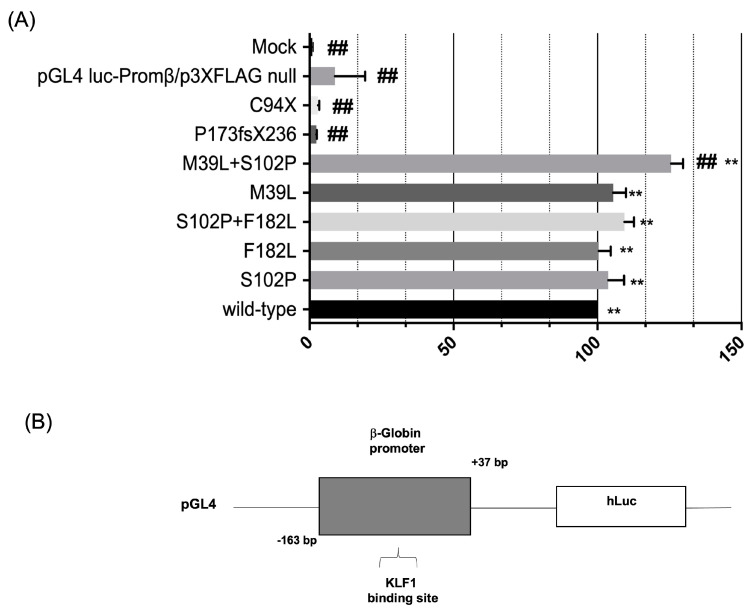
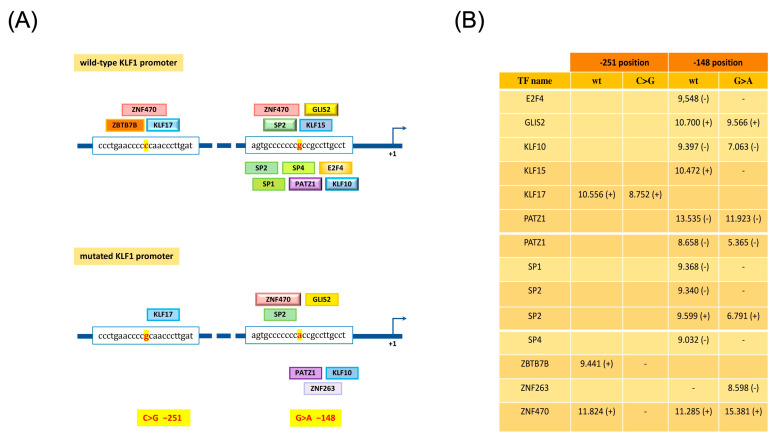
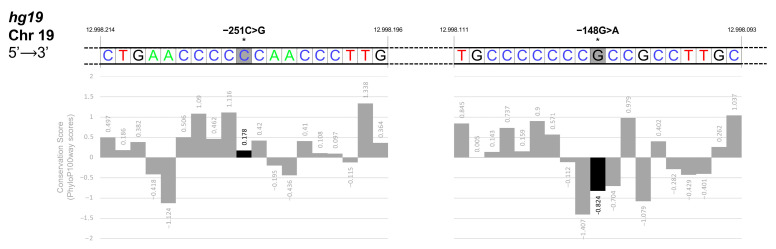
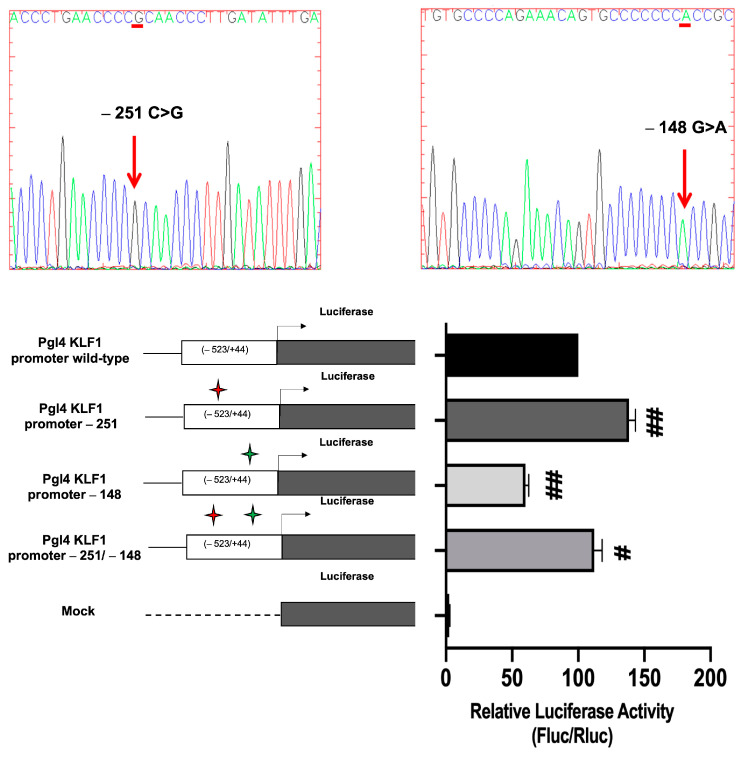
The erythroid transcriptional factor Krüppel-like factor 1 (KLF1) is a master regulator of erythropoiesis. Mutations that cause KLF1 haploinsufficiency have been linked to increased fetal hemoglobin (HbF) and hemoglobin A2 (HbA2) levels with ameliorative effects on the severity of β-thalassemia. With the aim of determining if KLF1 gene variations might play a role in the modulation of β-thalassemia, in this study we screened 17 subjects showing a β-thalassemia-like phenotype with a slight or marked increase in HbA2 and HbF levels. Overall, seven KLF1 gene variants were identified, of which two were novel. Functional studies were performed in K562 cells to clarify the pathogenic significance of these mutations. Our study confirmed the ameliorative effect on the thalassemia phenotype for some of these variants but also raised the notion that certain mutations may have deteriorating effects by increasing KLF1 expression levels or enhancing its transcriptional activity. Our results indicate that functional studies are required to evaluate the possible effects of KLF1 mutations, particularly in the case of the co-existence of two or more mutations that could differently contribute to KLF1 expression or transcriptional activity and consequently to the thalassemia phenotype.
Keywords: HbA2; HbF; KLF1; gene expression; globin gene switching; thalassemia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Differential role of Kruppel like factor 1 (KLF1) gene in red blood cell disorders.Genomics. 2019 Dec;111(6):1771-1776. doi: 10.1016/j.ygeno.2018.11.032. Epub 2018 Dec 5. Genomics. 2019. PMID: 30529538
-
Genetic variation of Krüppel-like factor 1 (KLF1) and fetal hemoglobin (HbF) levels in β0-thalassemia/HbE disease.Int J Hematol. 2018 Mar;107(3):297-310. doi: 10.1007/s12185-017-2357-3. Epub 2017 Oct 24. Int J Hematol. 2018. PMID: 29067594
-
Changes in HbA2 and HbF in alpha thalassemia carriers with KLF1 mutation.Blood Cells Mol Dis. 2017 May;64:30-32. doi: 10.1016/j.bcmd.2017.03.007. Epub 2017 Mar 18. Blood Cells Mol Dis. 2017. PMID: 28342932
-
Three fingers on the switch: Krüppel-like factor 1 regulation of γ-globin to β-globin gene switching.Curr Opin Hematol. 2013 May;20(3):193-200. doi: 10.1097/MOH.0b013e32835f59ba. Curr Opin Hematol. 2013. PMID: 23474875 Review.
-
Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis.Cells. 2022 Sep 29;11(19):3069. doi: 10.3390/cells11193069. Cells. 2022. PMID: 36231031 Free PMC article. Review.
Cited by
-
Erythroid Krüppel-Like Factor (KLF1): A Surprisingly Versatile Regulator of Erythroid Differentiation.Adv Exp Med Biol. 2024;1459:217-242. doi: 10.1007/978-3-031-62731-6_10. Adv Exp Med Biol. 2024. PMID: 39017846 Free PMC article. Review.
-
Trans-acting genetic modifiers of clinical severity in heterozygous β-Thalassemia trait.Ann Hematol. 2024 Nov;103(11):4437-4447. doi: 10.1007/s00277-024-06007-0. Epub 2024 Sep 24. Ann Hematol. 2024. PMID: 39316111 Review.
-
Chemical Profile and Promising Applications of Cucurbita pepo L. Flowers.Antioxidants (Basel). 2024 Nov 30;13(12):1476. doi: 10.3390/antiox13121476. Antioxidants (Basel). 2024. PMID: 39765805 Free PMC article.
References
-
- De Martino M., Sessa R., Rosaria Storino M., Giuliano M., Trombetti S., Catapano R., Lo Bianco A., Izzo P., Grosso M. Transcriptional Repressors of Fetal Globin Genes as Novel Therapeutic Targets in Beta-Thalassemia. In: Zakaria M., Hassan T., editors. Beta Thalassemia. IntechOpen; London, UK: 2020.
LinkOut - more resources
Full Text Sources
Miscellaneous
