

PGE2 Produced by Exogenous MSCs Promotes Immunoregulation in ARDS Induced by Highly Pathogenic Influenza A through Activation of the Wnt-β-Catenin Signaling Pathway
- PMID: 37108459
- PMCID: PMC10138595
- DOI: 10.3390/ijms24087299
PGE2 Produced by Exogenous MSCs Promotes Immunoregulation in ARDS Induced by Highly Pathogenic Influenza A through Activation of the Wnt-β-Catenin Signaling Pathway
Abstract
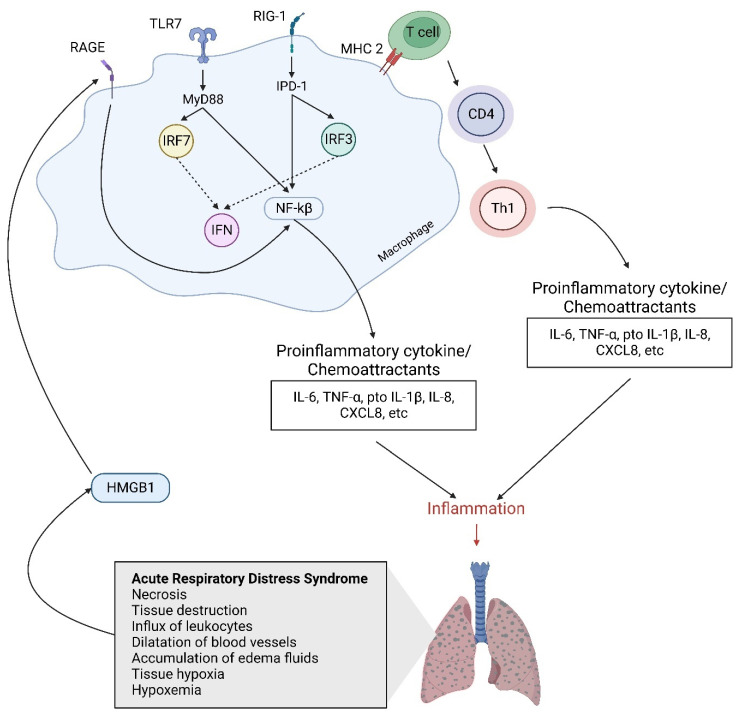
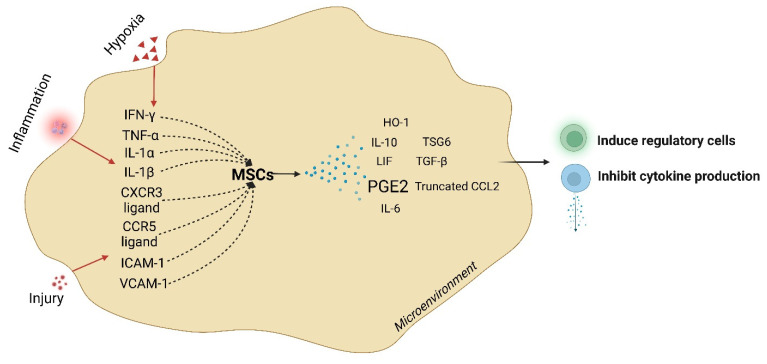
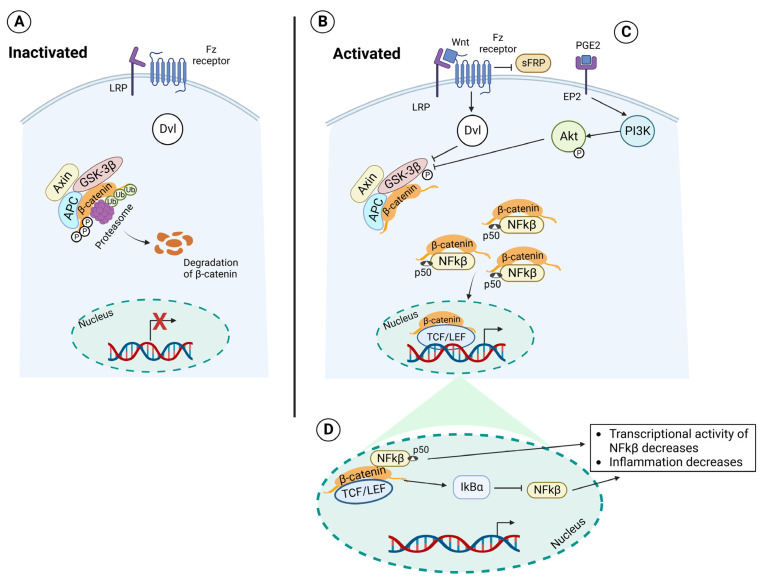
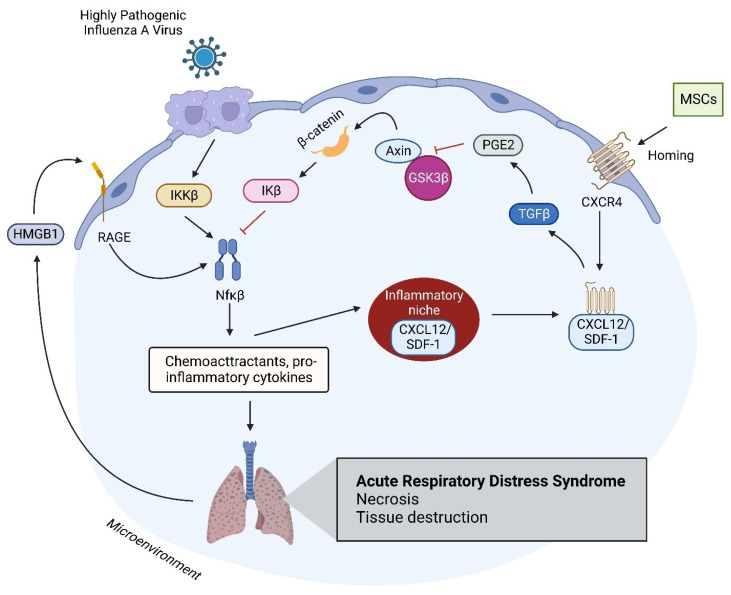
Acute respiratory distress syndrome is an acute respiratory failure caused by cytokine storms; highly pathogenic influenza A virus infection can induce cytokine storms. The innate immune response is vital in this cytokine storm, acting by activating the transcription factor NF-κB. Tissue injury releases a danger-associated molecular pattern that provides positive feedback for NF-κB activation. Exogenous mesenchymal stem cells can also modulate immune responses by producing potent immunosuppressive substances, such as prostaglandin E2. Prostaglandin E2 is a critical mediator that regulates various physiological and pathological processes through autocrine or paracrine mechanisms. Activation of prostaglandin E2 results in the accumulation of unphosphorylated β-catenin in the cytoplasm, which subsequently reaches the nucleus to inhibit the transcription factor NF-κB. The inhibition of NF-κB by β-catenin is a mechanism that reduces inflammation.
Keywords: NF-κB; PGE2; acute respiratory distress syndrome; infectious disease; influenza virus; mesenchymal stem cell; β-catenin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Wnt/β-catenin signaling pathway inhibits porcine reproductive and respiratory syndrome virus replication by enhancing the nuclear factor-κB-dependent innate immune response.Vet Microbiol. 2020 Dec;251:108904. doi: 10.1016/j.vetmic.2020.108904. Epub 2020 Oct 23. Vet Microbiol. 2020. PMID: 33181435
-
Activation of Wnt/β-catenin signalling promotes mesenchymal stem cells to repair injured alveolar epithelium induced by lipopolysaccharide in mice.Stem Cell Res Ther. 2015 Apr 11;6(1):65. doi: 10.1186/s13287-015-0060-y. Stem Cell Res Ther. 2015. Retraction in: Stem Cell Res Ther. 2022 Feb 3;13(1):50. doi: 10.1186/s13287-022-02731-4. PMID: 25889393 Free PMC article. Retracted.
-
Wnt/beta-catenin signaling regulates cytokine-induced human inducible nitric oxide synthase expression by inhibiting nuclear factor-kappaB activation in cancer cells.Cancer Res. 2009 May 1;69(9):3764-71. doi: 10.1158/0008-5472.CAN-09-0014. Epub 2009 Apr 21. Cancer Res. 2009. PMID: 19383900
-
Interplay of Opposing Effects of the WNT/β-Catenin Pathway and PPARγ and Implications for SARS-CoV2 Treatment.Front Immunol. 2021 Apr 13;12:666693. doi: 10.3389/fimmu.2021.666693. eCollection 2021. Front Immunol. 2021. PMID: 33927728 Free PMC article. Review.
-
Potential role of nutraceuticals via targeting a Wnt/β-catenin and NF-κB pathway in treatment of osteoarthritis.J Food Biochem. 2022 Dec;46(12):e14427. doi: 10.1111/jfbc.14427. Epub 2022 Sep 27. J Food Biochem. 2022. PMID: 36165556 Review.
Cited by
-
Protective effect of astaxanthin on chronic prostatitis/chronic pelvic pain syndrome in rat through modulating NF-κB signaling pathway.Transl Androl Urol. 2024 Sep 30;13(9):1971-1983. doi: 10.21037/tau-24-190. Epub 2024 Sep 26. Transl Androl Urol. 2024. PMID: 39434738 Free PMC article.
-
Mechanism of action of miR-15a-5p and miR-152-3p in paraquat-induced pulmonary fibrosis through Wnt/β-catenin signaling mediation.PeerJ. 2024 Jul 8;12:e17662. doi: 10.7717/peerj.17662. eCollection 2024. PeerJ. 2024. PMID: 38993979 Free PMC article. Review.
-
Dishevelled Has Anti-Viral Activity in Rift Valley Fever Virus Infected Aedes aegypti.Viruses. 2023 Oct 24;15(11):2140. doi: 10.3390/v15112140. Viruses. 2023. PMID: 38005818 Free PMC article.
-
Biomarkers of pediatric Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis through single-cell transcriptomics.Nat Commun. 2025 Jul 25;16(1):6888. doi: 10.1038/s41467-025-62090-5. Nat Commun. 2025. PMID: 40715093 Free PMC article.
-
Apoptotic vesicles rescue impaired mesenchymal stem cells and their therapeutic capacity for osteoporosis by restoring miR-145a-5p deficiency.J Nanobiotechnology. 2024 Sep 20;22(1):580. doi: 10.1186/s12951-024-02829-2. J Nanobiotechnology. 2024. PMID: 39304875 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
