

Autophagy in Spinocerebellar Ataxia Type 3: From Pathogenesis to Therapeutics
- PMID: 37108570
- PMCID: PMC10138583
- DOI: 10.3390/ijms24087405
Autophagy in Spinocerebellar Ataxia Type 3: From Pathogenesis to Therapeutics
Abstract
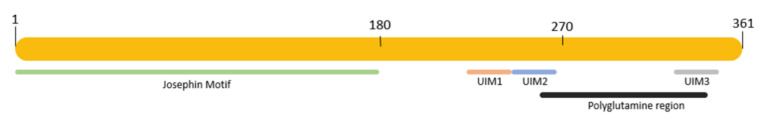
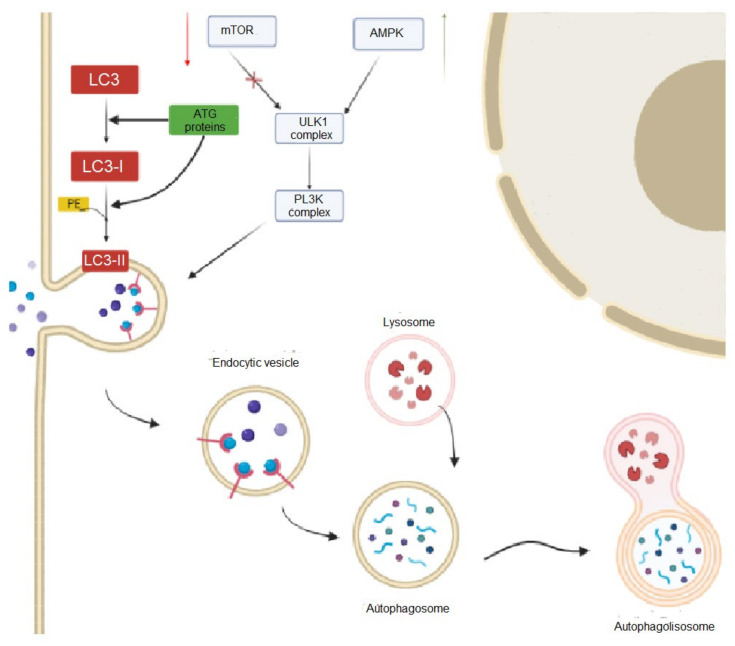
Machado-Joseph disease (MJD) or spinocerebellar ataxia 3 (SCA3) is a rare, inherited, monogenic, neurodegenerative disease, and the most common SCA worldwide. MJD/SCA3 causative mutation is an abnormal expansion of the triplet CAG at exon 10 within the ATXN3 gene. The gene encodes for ataxin-3, which is a deubiquitinating protein that is also involved in transcriptional regulation. In normal conditions, the ataxin-3 protein polyglutamine stretch has between 13 and 49 glutamines. However, in MJD/SCA3 patients, the size of the stretch increases from 55 to 87, contributing to abnormal protein conformation, insolubility, and aggregation. The formation of aggregates, which is a hallmark of MJD/SCA3, compromises different cell pathways, leading to an impairment of cell clearance mechanisms, such as autophagy. MJD/SCA3 patients display several signals and symptoms in which the most prominent is ataxia. Neuropathologically, the regions most affected are the cerebellum and the pons. Currently, there are no disease-modifying therapies, and patients rely only on supportive and symptomatic treatments. Due to these facts, there is a huge research effort to develop therapeutic strategies for this incurable disease. This review aims to bring together current state-of-the-art strategies regarding the autophagy pathway in MJD/SCA3, focusing on evidence for its impairment in the disease context and, importantly, its targeting for the development of pharmacological and gene-based therapies.
Keywords: Machado–Joseph disease; ataxin-3; autophagy; neurodegeneration; spinocerebellar ataxia type 3.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
