

Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies
- PMID: 37108642
- PMCID: PMC10139437
- DOI: 10.3390/ijms24087481
Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies
Abstract
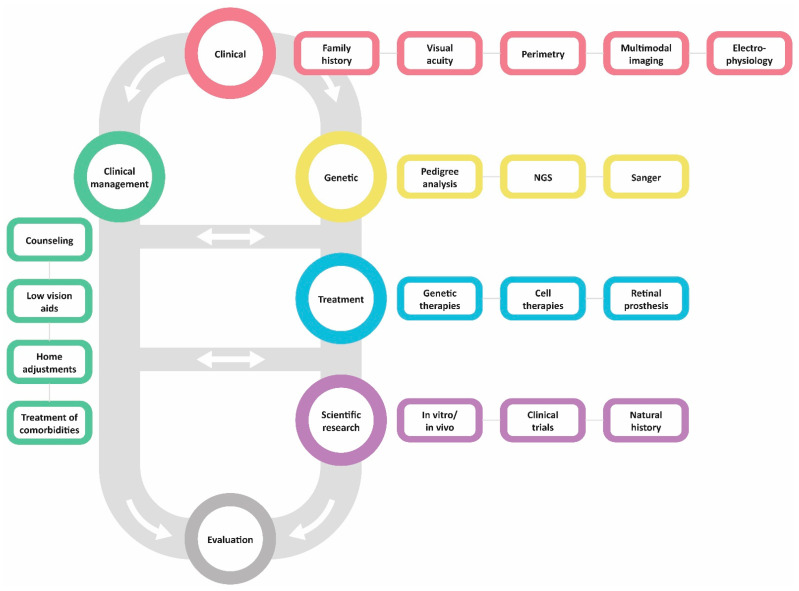
Retinitis pigmentosa (RP) comprises a group of inherited retinal dystrophies characterized by the degeneration of rod photoreceptors, followed by the degeneration of cone photoreceptors. As a result of photoreceptor degeneration, affected individuals experience gradual loss of visual function, with primary symptoms of progressive nyctalopia, constricted visual fields and, ultimately, central vision loss. The onset, severity and clinical course of RP shows great variability and unpredictability, with most patients already experiencing some degree of visual disability in childhood. While RP is currently untreatable for the majority of patients, significant efforts have been made in the development of genetic therapies, which offer new hope for treatment for patients affected by inherited retinal dystrophies. In this exciting era of emerging gene therapies, it remains imperative to continue supporting patients with RP using all available options to manage their condition. Patients with RP experience a wide variety of physical, mental and social-emotional difficulties during their lifetime, of which some require timely intervention. This review aims to familiarize readers with clinical management options that are currently available for patients with RP.
Keywords: clinical management; gene therapy; genetic counseling; genetics; low vision; low-vision rehabilitation; retinitis pigmentosa.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
