

Genetic Characteristics and Phylogeographic Dynamics of Lagoviruses, 1988-2021
- PMID: 37112796
- PMCID: PMC10143477
- DOI: 10.3390/v15040815
Genetic Characteristics and Phylogeographic Dynamics of Lagoviruses, 1988-2021
Abstract
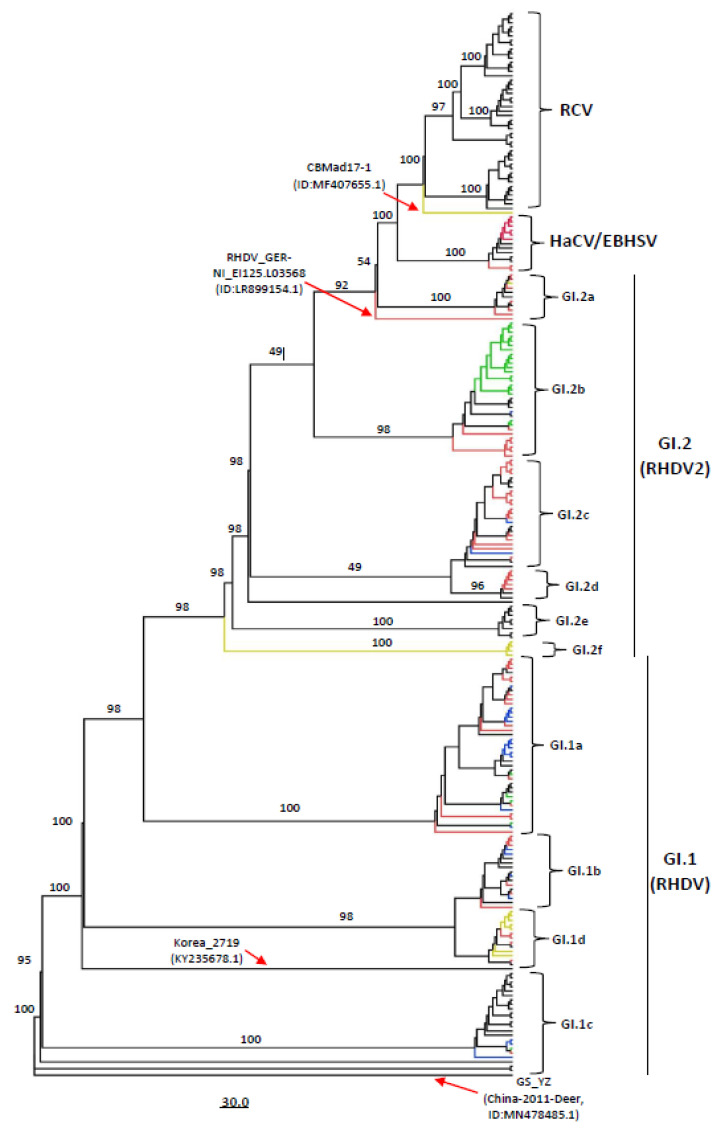
Rabbit haemorrhagic disease virus (RHDV), European brown hare syndrome virus (EBHSV), rabbit calicivirus (RCV), and hare calicivirus (HaCV) belong to the genus Lagovirus of the Caliciviridae family that causes severe diseases in rabbits and several hare (Lepus) species. Previously, Lagoviruses were classified into two genogroups, e.g., GI (RHDVs and RCVs) and GII (EBHSV and HaCV) based on partial genomes, e.g., VP60 coding sequences. Herein, we provide a robust phylogenetic classification of all the Lagovirus strains based on full-length genomes, grouping all the available 240 strains identified between 1988 and 2021 into four distinct clades, e.g., GI.1 (classical RHDV), GI.2 (RHDV2), HaCV/EBHSV, and RCV, where the GI.1 clade is further classified into four (GI.1a-d) and GI.2 into six sub-clades (GI.2a-f). Moreover, the phylogeographic analysis revealed that the EBHSV and HaCV strains share their ancestor with the GI.1, while the RCV shares with the GI.2. In addition, all 2020-2021 RHDV2 outbreak strains in the USA are connected to the strains from Canada and Germany, while RHDV strains isolated in Australia are connected with the USA-Germany haplotype RHDV strain. Furthermore, we identified six recombination events in the VP60, VP10, and RNA-dependent RNA polymerase (RdRp) coding regions using the full-length genomes. The amino acid variability analysis showed that the variability index exceeded the threshold of 1.00 in the ORF1-encoded polyprotein and ORF2-encoded VP10 protein, respectively, indicating significant amino acid drift with the emergence of new strains. The current study is an update of the phylogenetic and phylogeographic information of Lagoviruses that may be used to map the evolutionary history and provide hints for the genetic basis of their emergence and re-emergence.
Keywords: amino acid variation; evolution; lagoviruses; phylogenetics; phylogeographic analysis; recombination.
Conflict of interest statement
Authors have declared no conflict of interest.
Figures





Comment in
-
Comment on Shah et al. Genetic Characteristics and Phylogeographic Dynamics of Lagoviruses, 1988-2021. Viruses 2023, 15, 815.Viruses. 2024 Jun 7;16(6):927. doi: 10.3390/v16060927. Viruses. 2024. PMID: 38932219 Free PMC article.
-
Reply to Abrantes et al. Recombination-Based Perspectives on Lagovirus Classification, Phylogenetic Patterns, and Evolutionary Dynamics. Comment on "Shah et al. Genetic Characteristics and Phylogeographic Dynamics of Lagoviruses, 1988-2021. Viruses 2023, 15, 815".Viruses. 2024 Jun 7;16(6):928. doi: 10.3390/v16060928. Viruses. 2024. PMID: 38932220 Free PMC article.
Similar articles
-
Recombination between non-structural and structural genes as a mechanism of selection in lagoviruses: The evolutionary dead-end of an RHDV2 isolated from European hare.Virus Res. 2024 Jan 2;339:199257. doi: 10.1016/j.virusres.2023.199257. Epub 2023 Nov 3. Virus Res. 2024. PMID: 38347757 Free PMC article.
-
Widespread occurrence of the non-pathogenic hare calicivirus (HaCV Lagovirus GII.2) in captive-reared and free-living wild hares in Europe.Transbound Emerg Dis. 2021 Mar;68(2):509-518. doi: 10.1111/tbed.13706. Epub 2020 Jul 14. Transbound Emerg Dis. 2021. PMID: 32603021 Free PMC article.
-
Potent Protease Inhibitors of Highly Pathogenic Lagoviruses: Rabbit Hemorrhagic Disease Virus and European Brown Hare Syndrome Virus.Microbiol Spectr. 2022 Aug 31;10(4):e0014222. doi: 10.1128/spectrum.00142-22. Epub 2022 Jun 29. Microbiol Spectr. 2022. PMID: 35766511 Free PMC article.
-
Characterisation of Lagovirus europaeus GI-RHDVs (Rabbit Haemorrhagic Disease Viruses) in Terms of Their Pathogenicity and Immunogenicity.Int J Mol Sci. 2024 May 14;25(10):5342. doi: 10.3390/ijms25105342. Int J Mol Sci. 2024. PMID: 38791380 Free PMC article. Review.
-
Lack of evidence for differences in the spread of classic (Lagovirus europaeus/GI.1) and novel (Lagovirus europaeus/GI.2) rabbit haemorrhagic disease viruses in Europe and North Africa.Vet Rec. 2022 Feb;190(3):e1067. doi: 10.1002/vetr.1067. Epub 2021 Oct 28. Vet Rec. 2022. PMID: 34713453 Review.
Cited by
-
Systematic Review of Phylogenetic Analysis Techniques for RNA Viruses Using Bioinformatics.Int J Mol Sci. 2025 Feb 28;26(5):2180. doi: 10.3390/ijms26052180. Int J Mol Sci. 2025. PMID: 40076803 Free PMC article.
-
Comment on Shah et al. Genetic Characteristics and Phylogeographic Dynamics of Lagoviruses, 1988-2021. Viruses 2023, 15, 815.Viruses. 2024 Jun 7;16(6):927. doi: 10.3390/v16060927. Viruses. 2024. PMID: 38932219 Free PMC article.
-
Reply to Abrantes et al. Recombination-Based Perspectives on Lagovirus Classification, Phylogenetic Patterns, and Evolutionary Dynamics. Comment on "Shah et al. Genetic Characteristics and Phylogeographic Dynamics of Lagoviruses, 1988-2021. Viruses 2023, 15, 815".Viruses. 2024 Jun 7;16(6):928. doi: 10.3390/v16060928. Viruses. 2024. PMID: 38932220 Free PMC article.
References
-
- Liu S.J., Xue H.P., Pu B.Q., Qian N.H. A new viral disease in rabbit. Anim. Husb. Vet. Med. 1984;16:253–255.
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous