

Understanding Mutations in Human SARS-CoV-2 Spike Glycoprotein: A Systematic Review & Meta-Analysis
- PMID: 37112836
- PMCID: PMC10142771
- DOI: 10.3390/v15040856
Understanding Mutations in Human SARS-CoV-2 Spike Glycoprotein: A Systematic Review & Meta-Analysis
Abstract
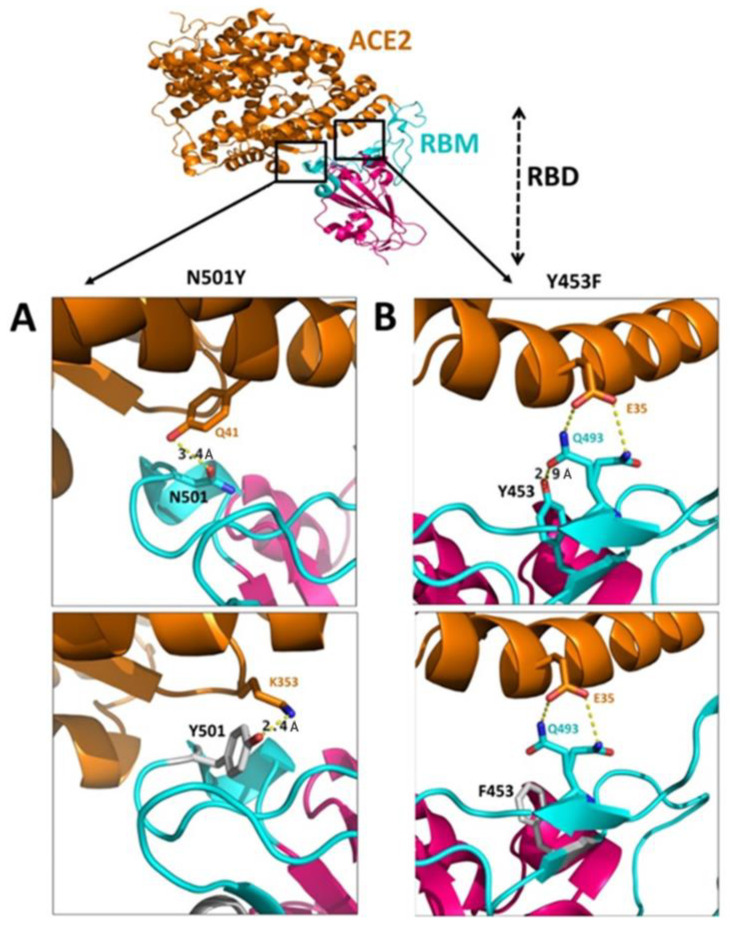
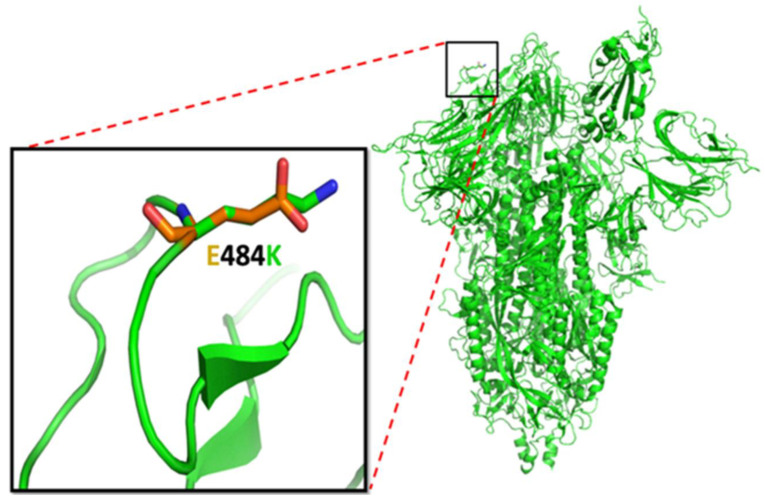
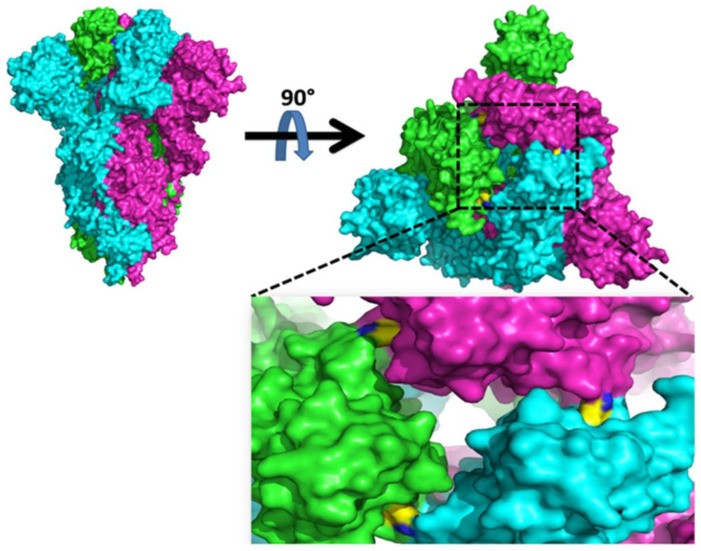
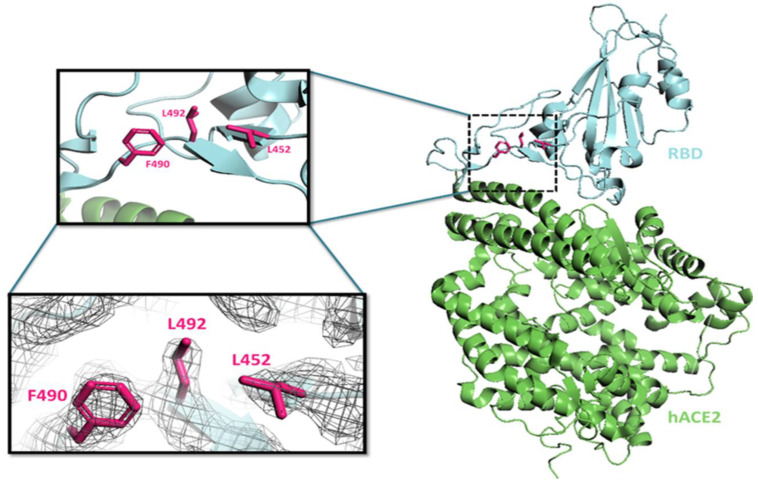
Genetic variant(s) of concern (VoC) of SARS-CoV-2 have been emerging worldwide due to mutations in the gene encoding spike glycoprotein. We performed comprehensive analyses of spike protein mutations in the significant variant clade of SARS-CoV-2, using the data available on the Nextstrain server. We selected various mutations, namely, A222V, N439K, N501Y, L452R, Y453F, E484K, K417N, T478K, L981F, L212I, N856K, T547K, G496S, and Y369C for this study. These mutations were chosen based on their global entropic score, emergence, spread, transmission, and their location in the spike receptor binding domain (RBD). The relative abundance of these mutations was mapped with global mutation D614G as a reference. Our analyses suggest the rapid emergence of newer global mutations alongside D614G, as reported during the recent waves of COVID-19 in various parts of the world. These mutations could be instrumentally imperative for the transmission, infectivity, virulence, and host immune system's evasion of SARS-CoV-2. The probable impact of these mutations on vaccine effectiveness, antigenic diversity, antibody interactions, protein stability, RBD flexibility, and accessibility to human cell receptor ACE2 was studied in silico. Overall, the present study can help researchers to design the next generation of vaccines and biotherapeutics to combat COVID-19 infection.
Keywords: COVID-19; SARS-CoV-2; VoC; evolution; mutations; spike protein.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
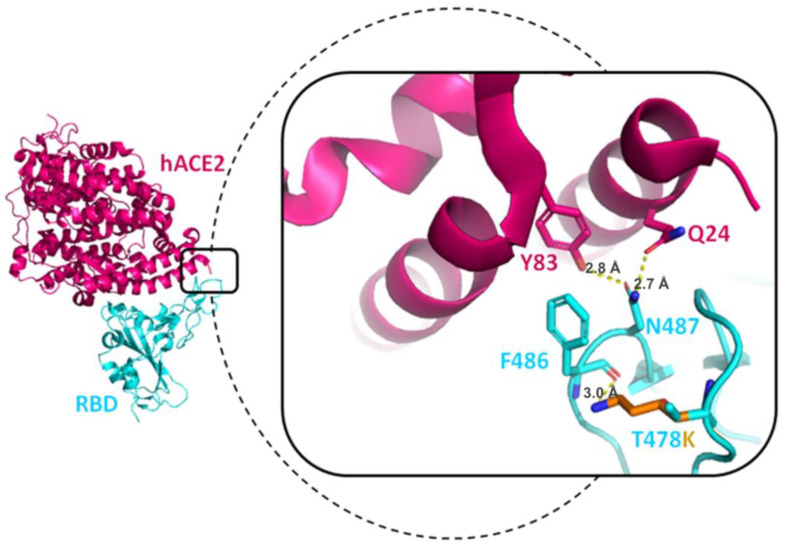
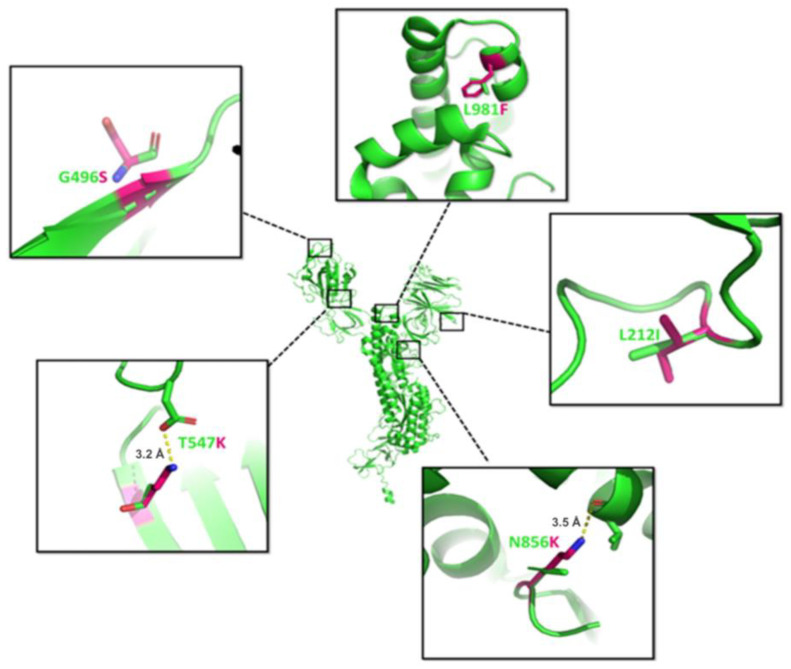
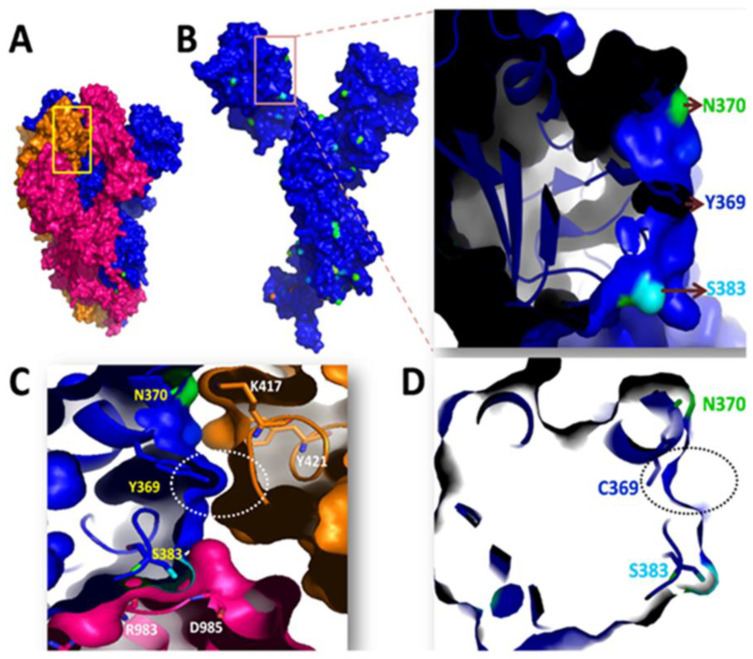
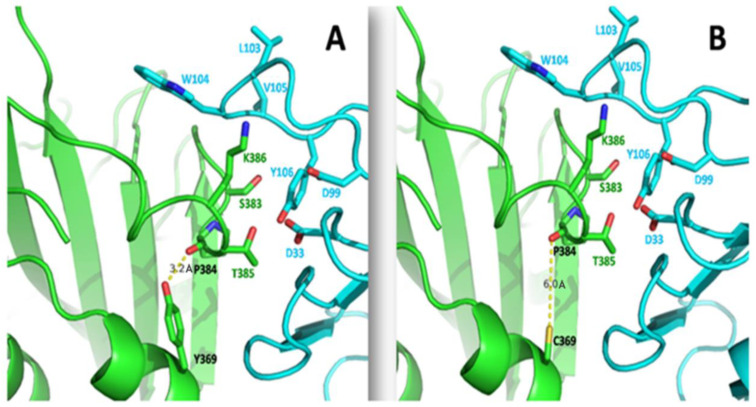
References
-
- WHO . Coronavirus Disease 2019 (COVID-19): Situation Report. Volume 51 WHO; Geneva, Switzerland: 2020.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
