

Safety and effectiveness of ataluren in patients with nonsense mutation DMD in the STRIDE Registry compared with the CINRG Duchenne Natural History Study (2015-2022): 2022 interim analysis
- PMID: 37115359
- PMCID: PMC10141820
- DOI: 10.1007/s00415-023-11687-1
Safety and effectiveness of ataluren in patients with nonsense mutation DMD in the STRIDE Registry compared with the CINRG Duchenne Natural History Study (2015-2022): 2022 interim analysis
Erratum in
-
Correction to: Safety and effectiveness of ataluren in patients with nonsense mutation DMD in the STRIDE Registry compared with the CINRG Duchenne Natural History Study (2015-2022): 2022 interim analysis.J Neurol. 2023 Sep;270(9):4583. doi: 10.1007/s00415-023-11864-2. J Neurol. 2023. PMID: 37460854 Free PMC article. No abstract available.
Abstract
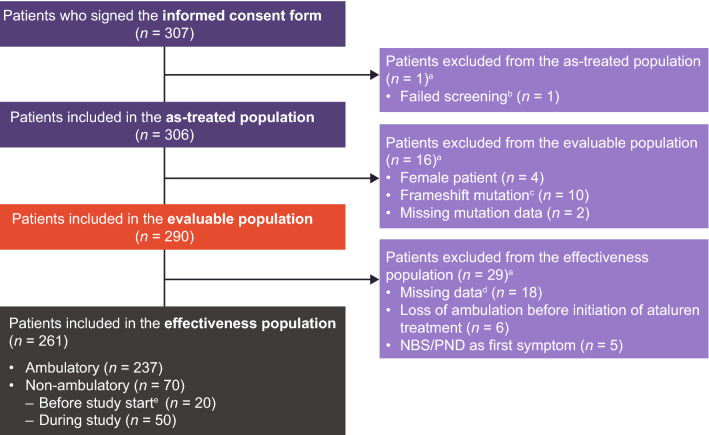
Objective: Strategic Targeting of Registries and International Database of Excellence (STRIDE) is an ongoing, international, multicenter registry of real-world ataluren use in individuals with nonsense mutation Duchenne muscular dystrophy (nmDMD) in clinical practice. This updated interim report (data cut-off: January 31, 2022), describes STRIDE patient characteristics and ataluren safety data, as well as the effectiveness of ataluren plus standard of care (SoC) in STRIDE versus SoC alone in the Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS).
Methods: Patients are followed up from enrollment for at least 5 years or until study withdrawal. Propensity score matching was performed to identify STRIDE and CINRG DNHS patients who were comparable in established predictors of disease progression.
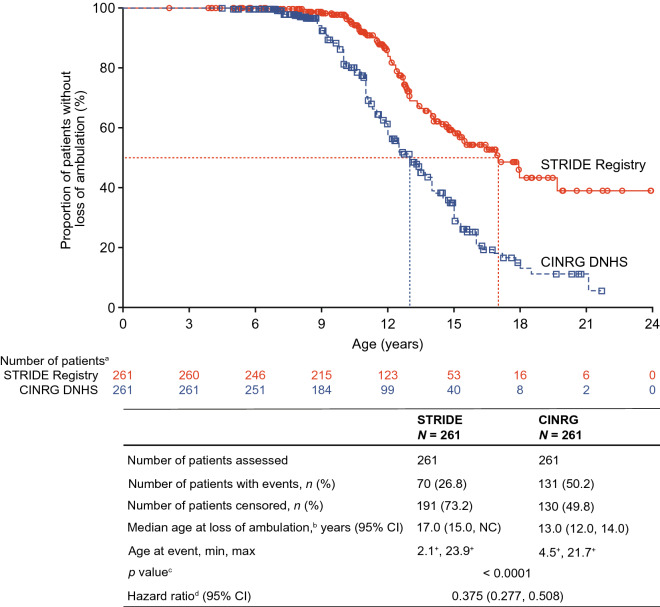
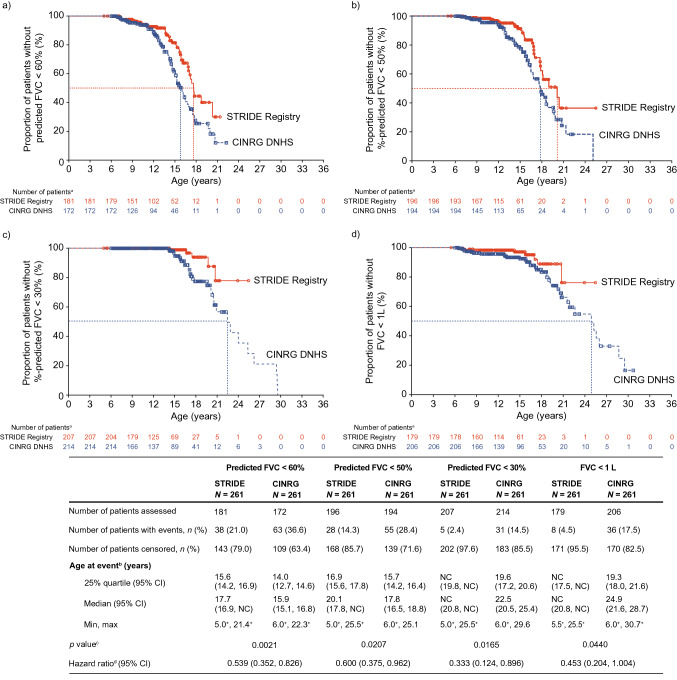
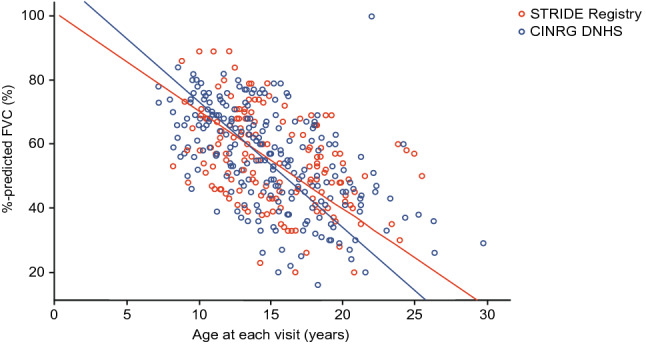
Results: As of January 31, 2022, 307 patients were enrolled from 14 countries. Mean (standard deviation [SD]) ages at first symptoms and at genetic diagnosis were 2.9 (1.7) years and 4.5 (3.7) years, respectively. Mean (SD) duration of ataluren exposure was 1671 (56.8) days. Ataluren had a favorable safety profile; most treatment-emergent adverse events were mild or moderate and unrelated to ataluren. Kaplan-Meier analyses demonstrated that ataluren plus SoC significantly delayed age at loss of ambulation by 4 years (p < 0.0001) and age at decline to %-predicted forced vital capacity of < 60% and < 50% by 1.8 years (p = 0.0021) and 2.3 years (p = 0.0207), respectively, compared with SoC alone.
Conclusion: Long-term, real-world treatment with ataluren plus SoC delays several disease progression milestones in individuals with nmDMD. NCT02369731; registration date: February 24, 2015.
Keywords: Ataluren; Effectiveness; Nonsense mutation Duchenne muscular dystrophy; STRIDE Registry; Safety.
© 2023. The Author(s).
Conflict of interest statement
EM has acted as an advisory board member for AveXis, Biogen, BioMarin, Bristol-Myers Squibb, Ionis Pharmaceuticals, Italfarmaco, Prosensa, PTC Therapeutics, Roche, Santhera Pharmaceuticals, Sarepta Therapeutics and Summit Therapeutics. ANO has received speaker and consultancy fees from PTC Therapeutics and is an investigator on clinical trials sponsored by Biogen, Italfarmaco, Roche, Sarepta Therapeutics and TAMDMD. FM has received consultancy fees from Akashi Therapeutics, Biogen, BioMarin, Catabasis, Italfarmaco, Pfizer, PTC Therapeutics, Roche, Sarepta Therapeutics and Tivorsan Pharmaceuticals, and is supported by University College London, London, UK, and the National Institute of Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children, National Health Service Foundation Trust. FB has received consultancy fees from PTC Therapeutics, Santhera Pharmaceuticals and Sarepta Therapeutics. ID has received consultancy fees from AveXis, Biogen, BioMarin and PTC Therapeutics. JK has acted as a consultant for Biogen, Novartis, Pfizer, PTC Therapeutics, Roche and Scholar Rock and has received research support for taking part in clinical research from Biogen, Novartis, PTC Therapeutics and Roche. MT has received lecture fees from Biogen and PTC Therapeutics; has acted as a consultant on DMD clinical trials for BioMarin, PTC Therapeutics, ReveraGen BioPharma and Sarepta Therapeutics; and has acted as an advisory board member for AveXis, Biogen and PTC Therapeutics. LM is a part owner of TRiNDS. HGD has served as a consultant for AGADA Biosciences, ReveraGen BioPharma and Solid GT, and is a cofounder and part owner of TRiNDS. SJ, AK and PT are full-time employees of PTC Therapeutics, Inc. CW is a full-time employee of PTC Therapeutics GmbH. EH has acted as a consultant on clinical trials of DMD for Capricor Therapeutics, Epirium Bio (formerly Cardero Therapeutics), Mallinckrodt Pharmaceuticals, Pfizer, PTC Therapeutics, Sanofi Genzyme, Santhera Pharmaceuticals and Sarepta Therapeutics. CM has served as a consultant for clinical trials with Astellas Pharma, Avidity Biosciences, BioMarin Pharmaceutical, Capricor Therapeutics, Catabasis Pharmaceuticals, Edgewise Therapeutics, Entrada Therapeutics, Epirium Bio (formerly Cardero Therapeutics), FibroGen, Hoffman La Roche, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals Sarepta Therapeutics and Solid Biosciences. He has received research support for clinical trials from Capricor Therapeutics, Catabasis Pharmaceuticals, Edgewise Therapeutics, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals and Sarepta Therapeutics. He serves on external advisory boards related to DMD for BioMarin Pharmaceutical, Capricor Therapeutics, Edgewise Therapeutics, Eli Lilly, PTC Therapeutics, Sarepta Therapeutics and Santhera Pharmaceuticals. MBDR reports no disclosures of interest.
Figures
References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
Research Materials
