

Genomic analysis unveils genome degradation events and gene flux in the emergence and persistence of S. Paratyphi A lineages
- PMID: 37115804
- PMCID: PMC10171690
- DOI: 10.1371/journal.ppat.1010650
Genomic analysis unveils genome degradation events and gene flux in the emergence and persistence of S. Paratyphi A lineages
Abstract
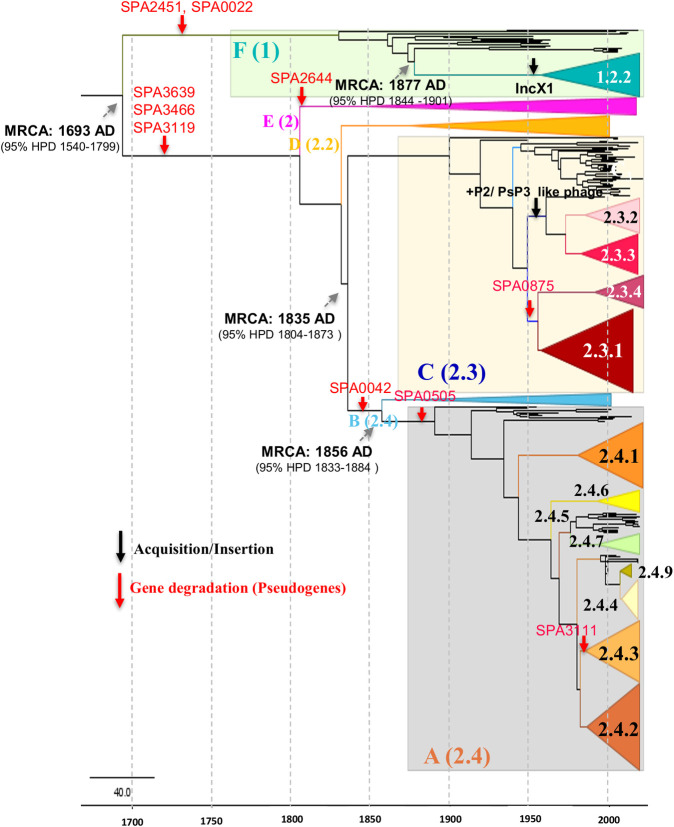
Paratyphoid fever caused by S. Paratyphi A is endemic in parts of South Asia and Southeast Asia. The proportion of enteric fever cases caused by S. Paratyphi A has substantially increased, yet only limited data is available on the population structure and genetic diversity of this serovar. We examined the phylogenetic distribution and evolutionary trajectory of S. Paratyphi A isolates collected as part of the Indian enteric fever surveillance study "Surveillance of Enteric Fever in India (SEFI)." In the study period (2017-2020), S. Paratyphi A comprised 17.6% (441/2503) of total enteric fever cases in India, with the isolates highly susceptible to all the major antibiotics used for treatment except fluoroquinolones. Phylogenetic analysis clustered the global S. Paratyphi A collection into seven lineages (A-G), and the present study isolates were distributed in lineages A, C and F. Our analysis highlights that the genome degradation events and gene acquisitions or losses are key molecular events in the evolution of new S. Paratyphi A lineages/sub-lineages. A total of 10 hypothetically disrupted coding sequences (HDCS) or pseudogenes-forming mutations possibly associated with the emergence of lineages were identified. The pan-genome analysis identified the insertion of P2/PSP3 phage and acquisition of IncX1 plasmid during the selection in 2.3.2/2.3.3 and 1.2.2 genotypes, respectively. We have identified six characteristic missense mutations associated with lipopolysaccharide (LPS) biosynthesis genes of S. Paratyphi A, however, these mutations confer only a low structural impact and possibly have minimal impact on vaccine effectiveness. Since S. Paratyphi A is human-restricted, high levels of genetic drift are not expected unless these bacteria transmit to naive hosts. However, public-health investigation and monitoring by means of genomic surveillance would be constantly needed to avoid S. Paratyphi A serovar becoming a public health threat similar to the S. Typhi of today.
Copyright: © 2023 Jacob et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Crump JA, Wain J. Salmonella. In Quah SR, Cockerham WC, editors, International Encyclopedia of Public Health. 2 ed. Elsevier. 2017. p. 425–433 10.1016/B978-0-12-803678-5.00394-5 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
