

TWIST2-mediated chromatin remodeling promotes fusion-negative rhabdomyosarcoma
- PMID: 37115930
- PMCID: PMC10146891
- DOI: 10.1126/sciadv.ade8184
TWIST2-mediated chromatin remodeling promotes fusion-negative rhabdomyosarcoma
Abstract
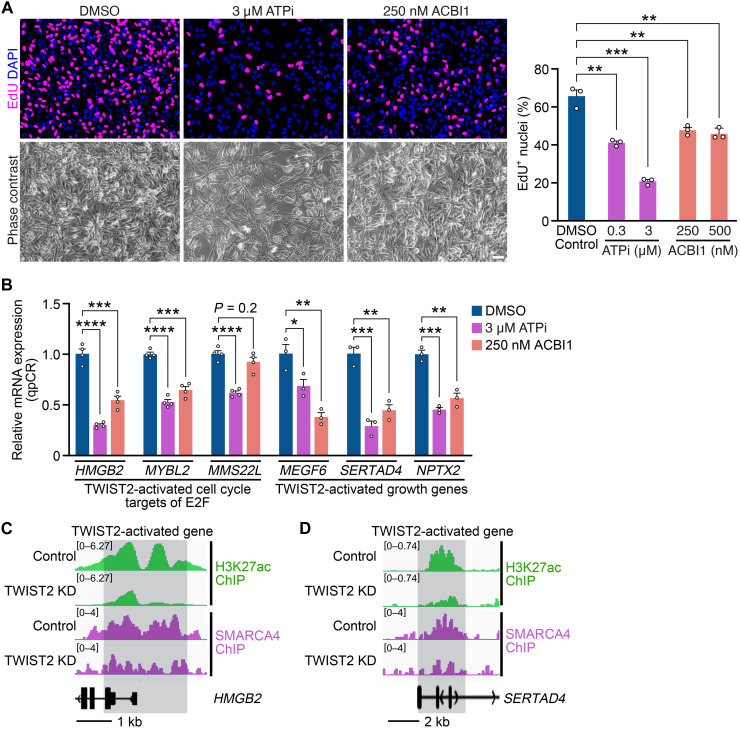
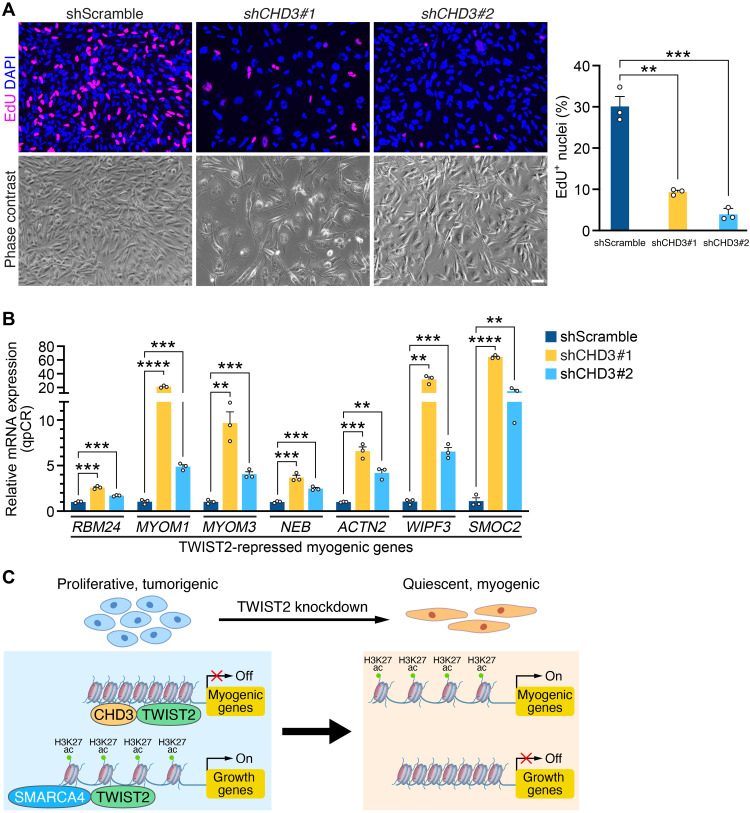
Rhabdomyosarcoma (RMS) is a common soft tissue sarcoma in children that resembles developing skeletal muscle. Unlike normal muscle cells, RMS cells fail to differentiate despite expression of the myogenic determination protein MYOD. The TWIST2 transcription factor is frequently overexpressed in fusion-negative RMS (FN-RMS). TWIST2 blocks differentiation by inhibiting MYOD activity in myoblasts, but its role in FN-RMS pathogenesis is incompletely understood. Here, we show that knockdown of TWIST2 enables FN-RMS cells to exit the cell cycle and undergo terminal myogenesis. TWIST2 knockdown also substantially reduces tumor growth in a mouse xenograft model of FN-RMS. Mechanistically, TWIST2 controls H3K27 acetylation at distal enhancers by interacting with the chromatin remodelers SMARCA4 and CHD3 to activate growth-related target genes and repress myogenesis-related target genes. These findings provide insights into the role of TWIST2 in maintaining an undifferentiated and tumorigenic state of FN-RMS and highlight the potential of suppressing TWIST2-regulated pathways to treat FN-RMS.
Figures
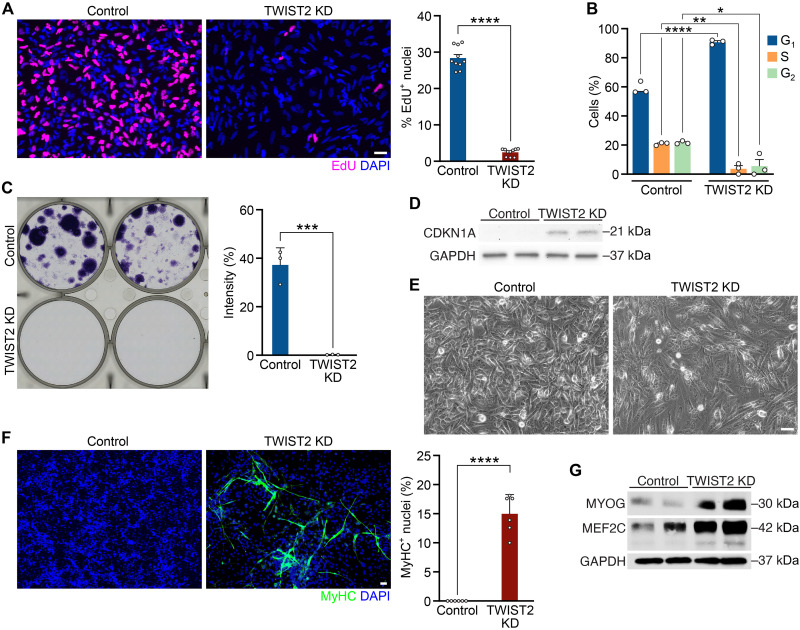
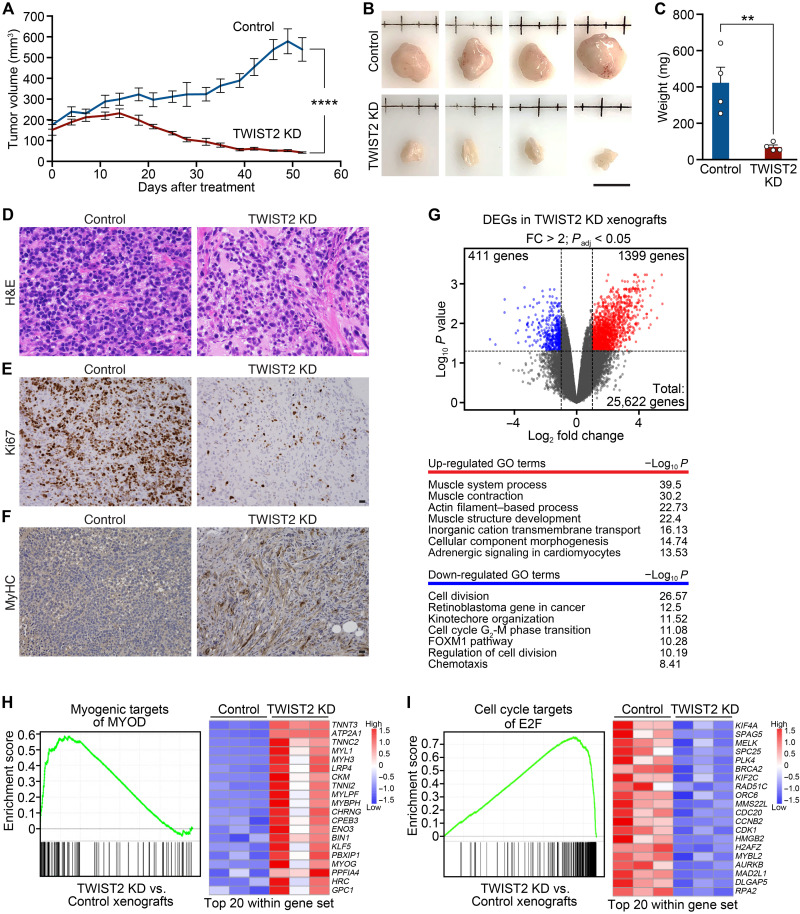
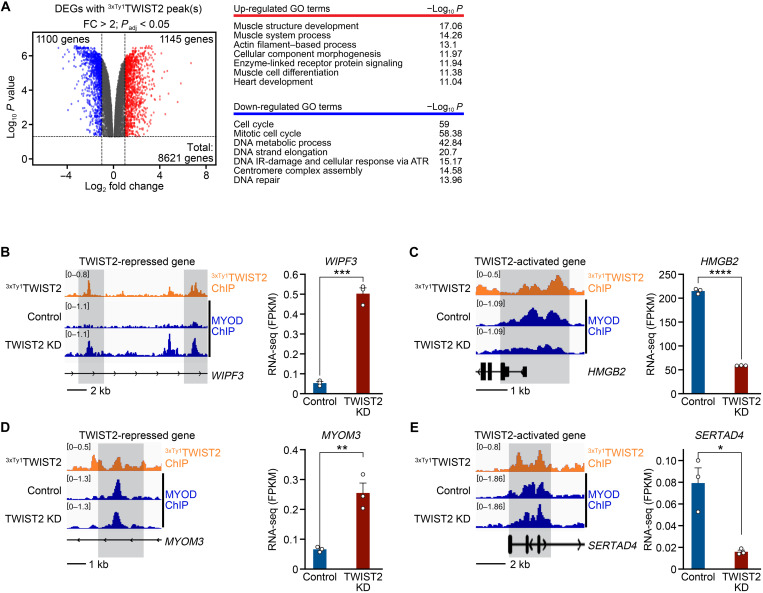

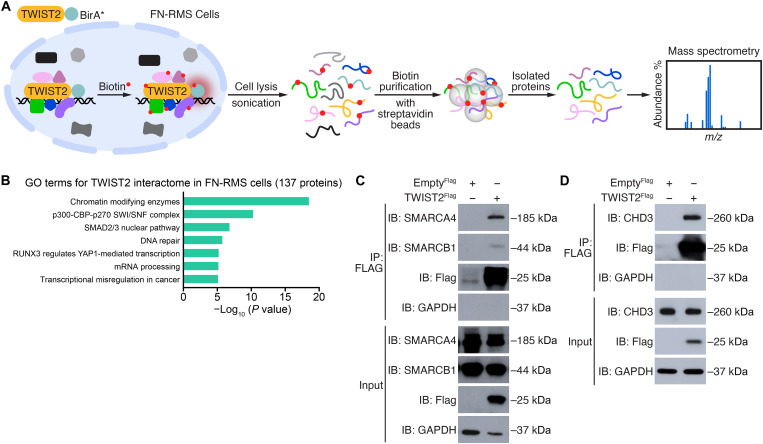
References
-
- M. Buckingham, P. W. J. Rigby, Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev. Cell 28, 225–238 (2014). - PubMed
-
- R. Saab, S. L. Spunt, S. X. Skapek, Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr. Top. Dev. Biol. 94, 197–234 (2011). - PubMed
-
- S. J. Tapscott, M. J. Thayer, H. Weintraub, Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science 259, 1450–1453 (1993). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
