

Toward an improved definition of a healthy microbiome for healthy aging
- PMID: 37118093
- PMCID: PMC10154212
- DOI: 10.1038/s43587-022-00306-9
Toward an improved definition of a healthy microbiome for healthy aging
Abstract
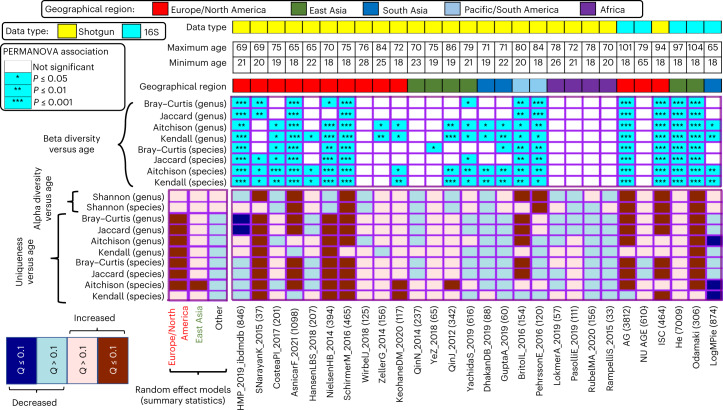
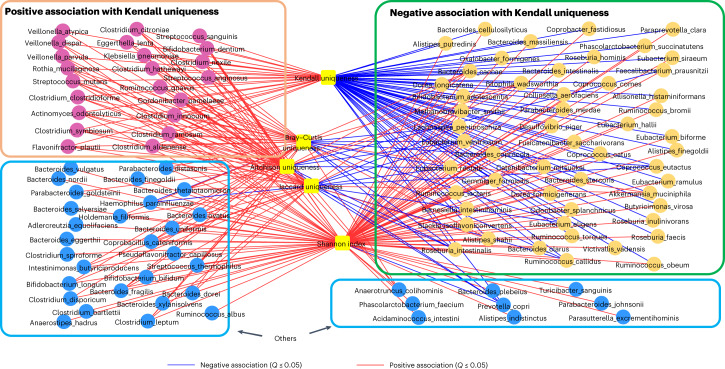
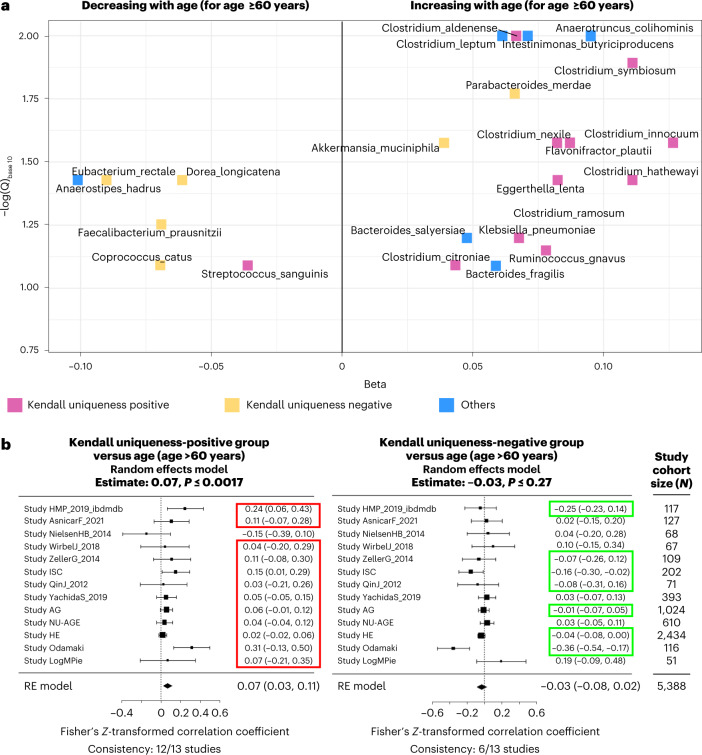
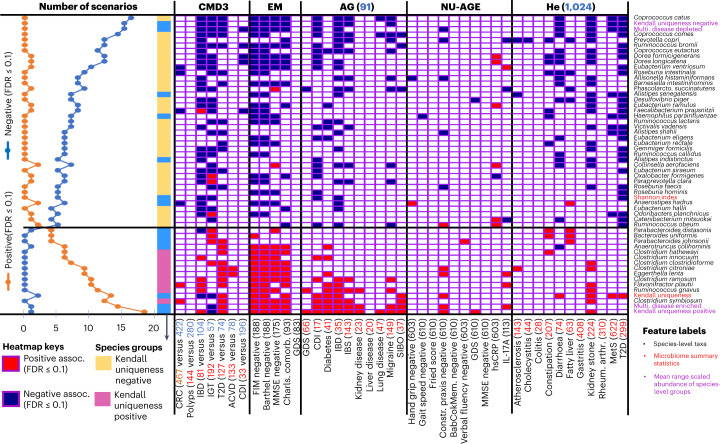
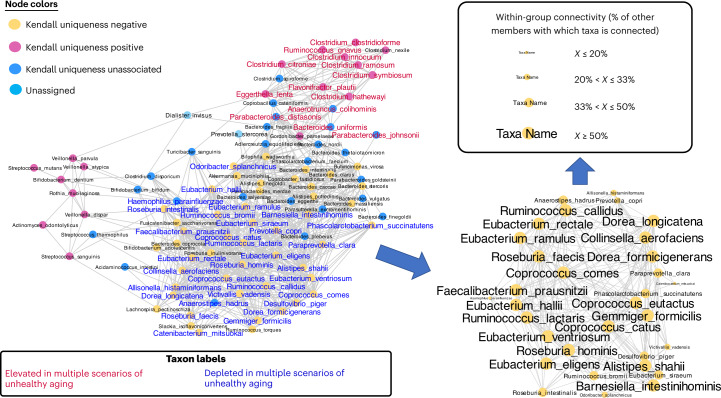
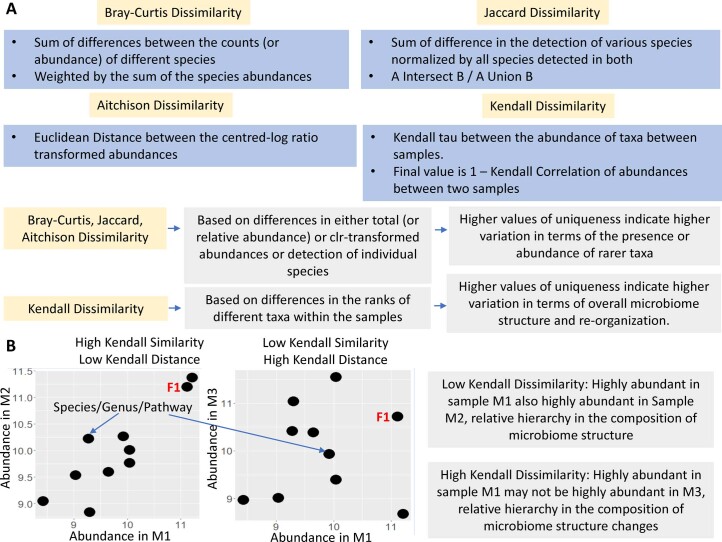
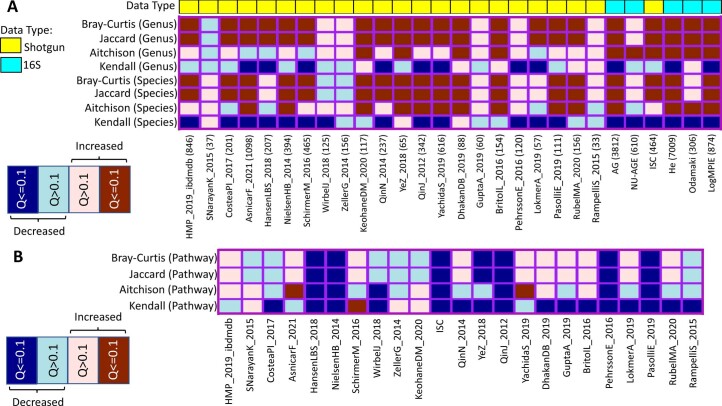
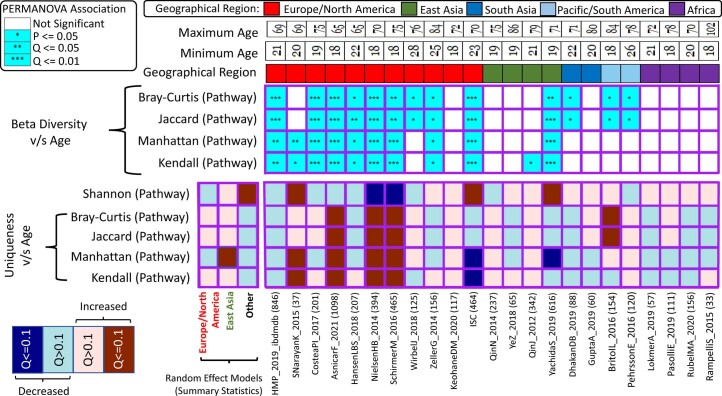
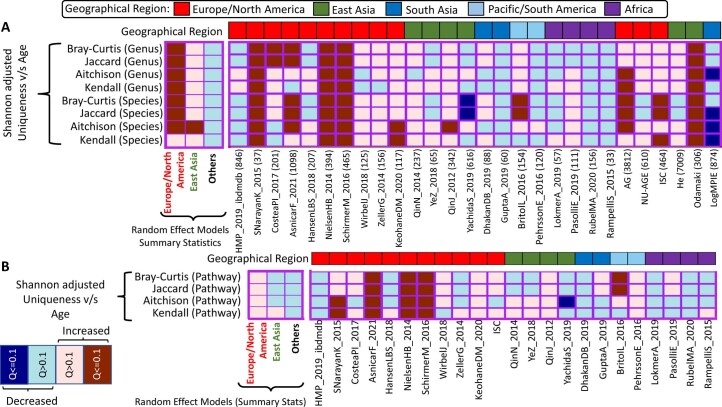
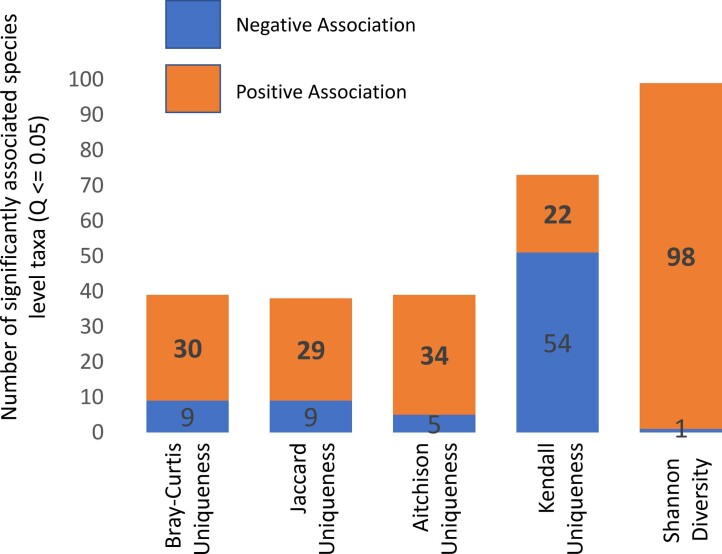
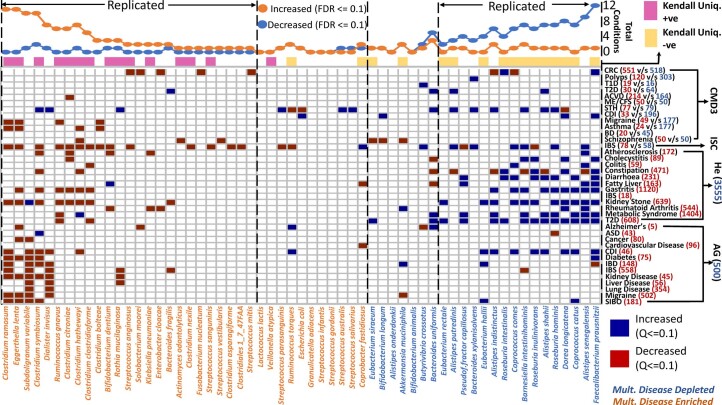
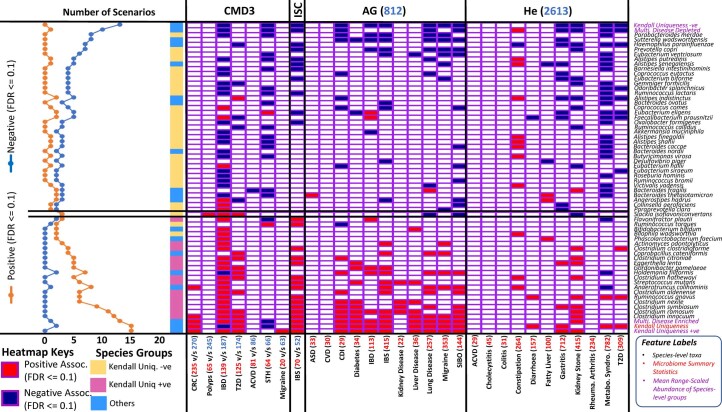
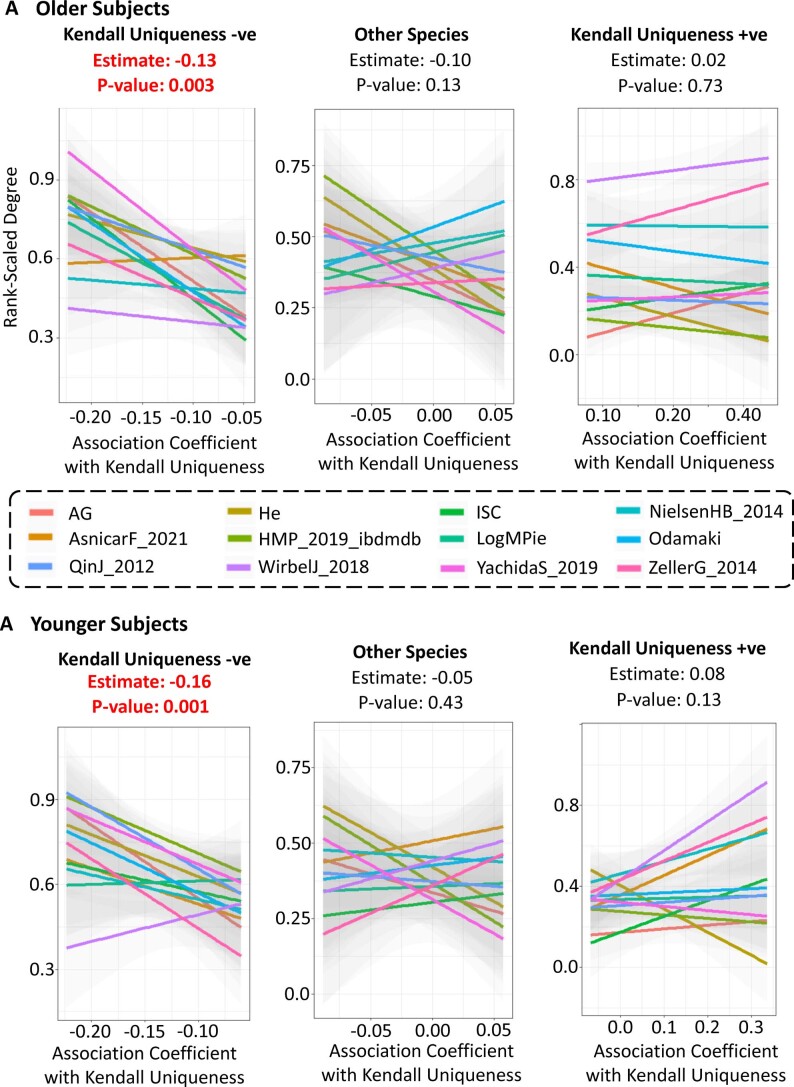
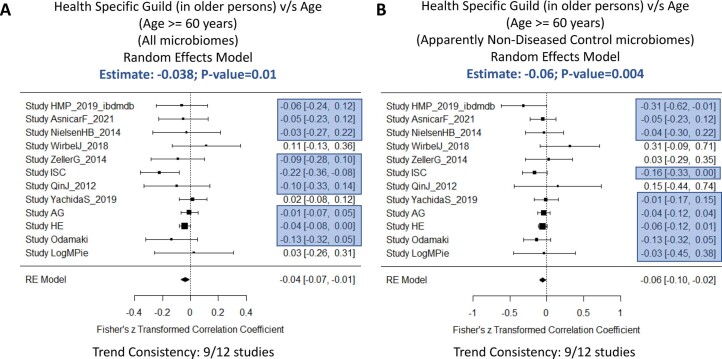
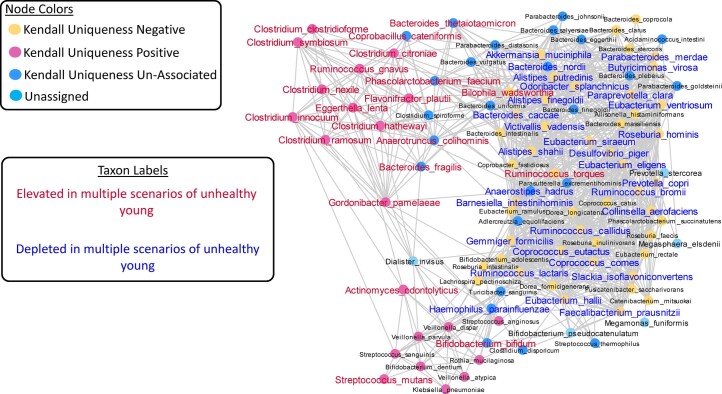
The gut microbiome is a modifier of disease risk because it interacts with nutrition, metabolism, immunity and infection. Aging-related health loss has been correlated with transition to different microbiome states. Microbiome summary indices including alpha diversity are apparently useful to describe these states but belie taxonomic differences that determine biological importance. We analyzed 21,000 fecal microbiomes from seven data repositories, across five continents spanning participant ages 18-107 years, revealing that microbiome diversity and uniqueness correlate with aging, but not healthy aging. Among summary statistics tested, only Kendall uniqueness accurately reflects loss of the core microbiome and the abundance and ranking of disease-associated and health-associated taxa. Increased abundance of these disease-associated taxa and depletion of a coabundant subset of health-associated taxa are a generic feature of aging. These alterations are stronger correlates of unhealthy aging than most microbiome summary statistics and thus help identify better targets for therapeutic modulation of the microbiome.
© 2022. The Author(s).
Conflict of interest statement
P.W.O.T. and T.S.G. are co-authors of a patent describing specific isolated bacterial strains and use of the composition for treating frailty and/or inflammation related to aging in older adults (applicant: University College Cork; inventors: O’Toole, Paul; Jeffery, Ian; Ghosh, Tarini S.; Tan, Huizi; Perez, Marta; Ntemiri, Alexandra; WO/2021/074431; pending). Seven of the taxa identified as being health-related in the current study are covered by the patent. F.S. has had a scientific advisory role to Kaleido Biosciences and Norgine. Those facts have had no bearing or constraint on the present paper.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
