

Heterochronic parabiosis reprograms the mouse brain transcriptome by shifting aging signatures in multiple cell types
- PMID: 37118429
- PMCID: PMC10154248
- DOI: 10.1038/s43587-023-00373-6
Heterochronic parabiosis reprograms the mouse brain transcriptome by shifting aging signatures in multiple cell types
Erratum in
-
Author Correction: Heterochronic parabiosis reprograms the mouse brain transcriptome by shifting aging signatures in multiple cell types.Nat Aging. 2025 Feb;5(2):333. doi: 10.1038/s43587-025-00804-6. Nat Aging. 2025. PMID: 39881191 Free PMC article. No abstract available.
Abstract
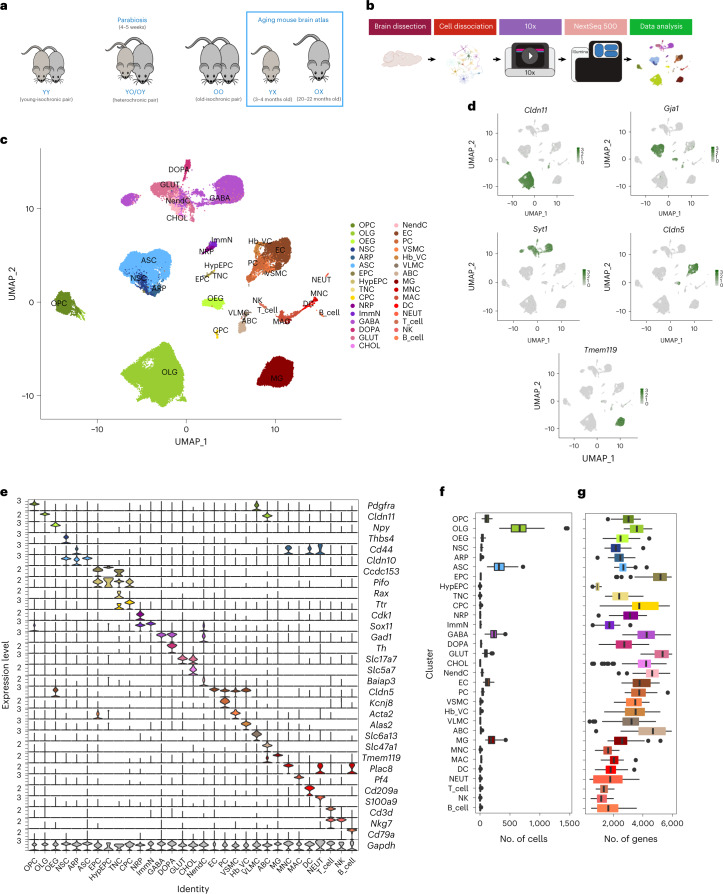
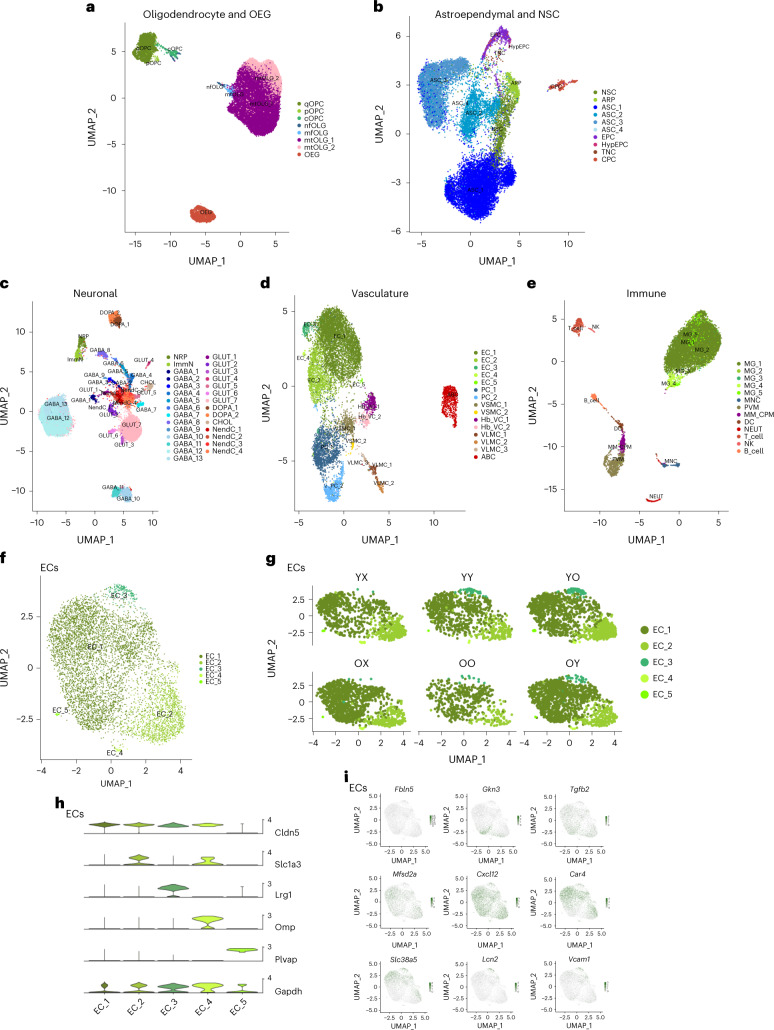
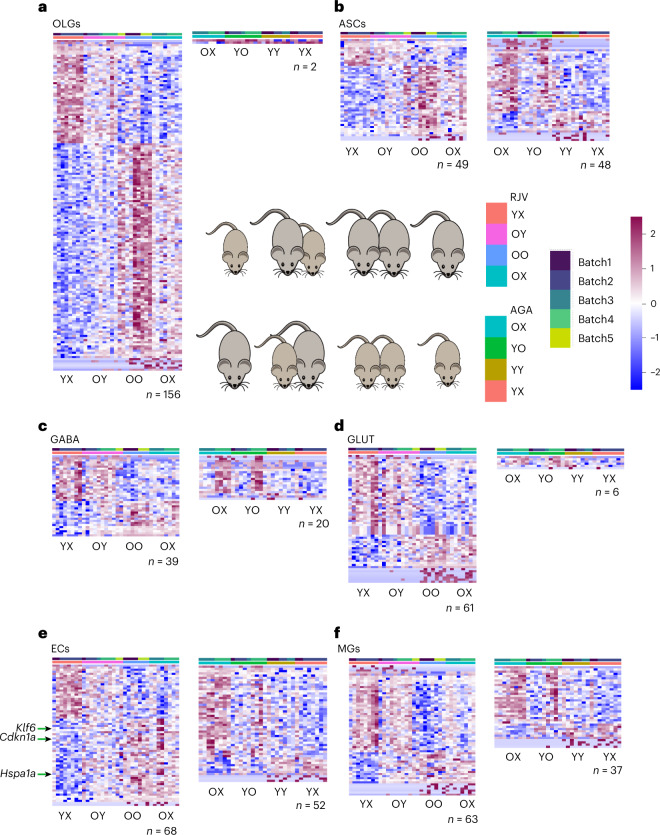
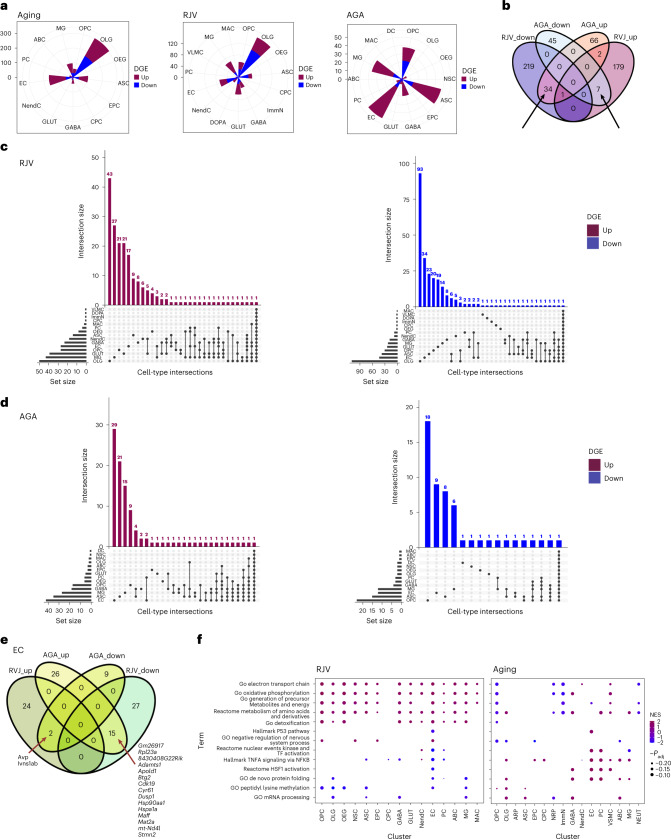
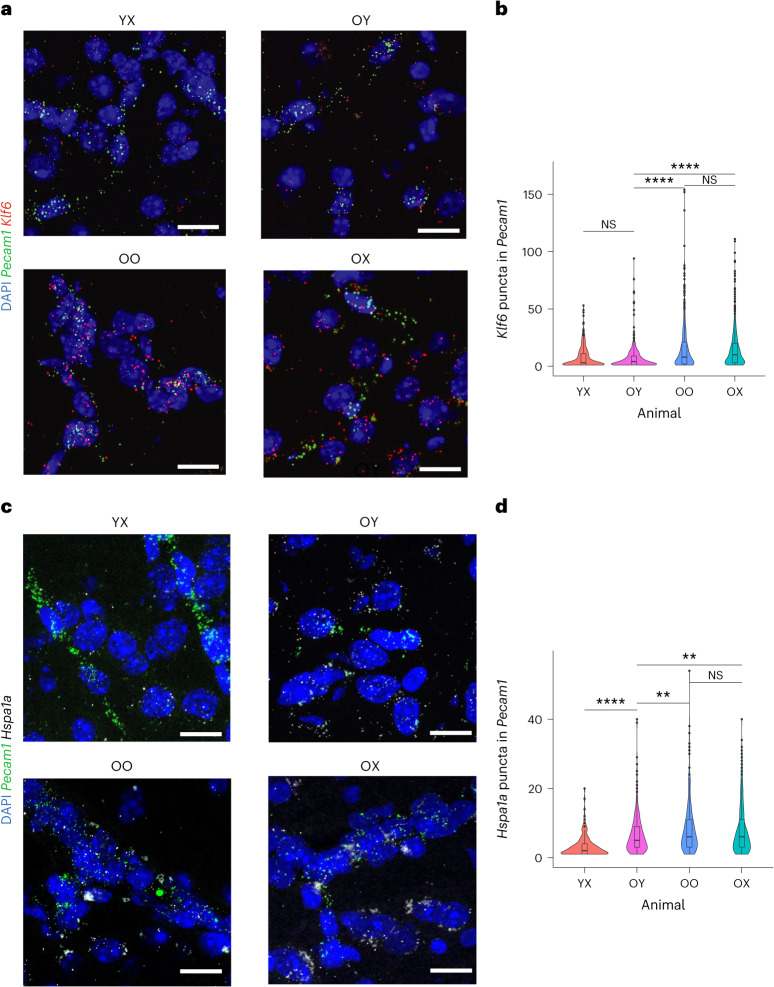
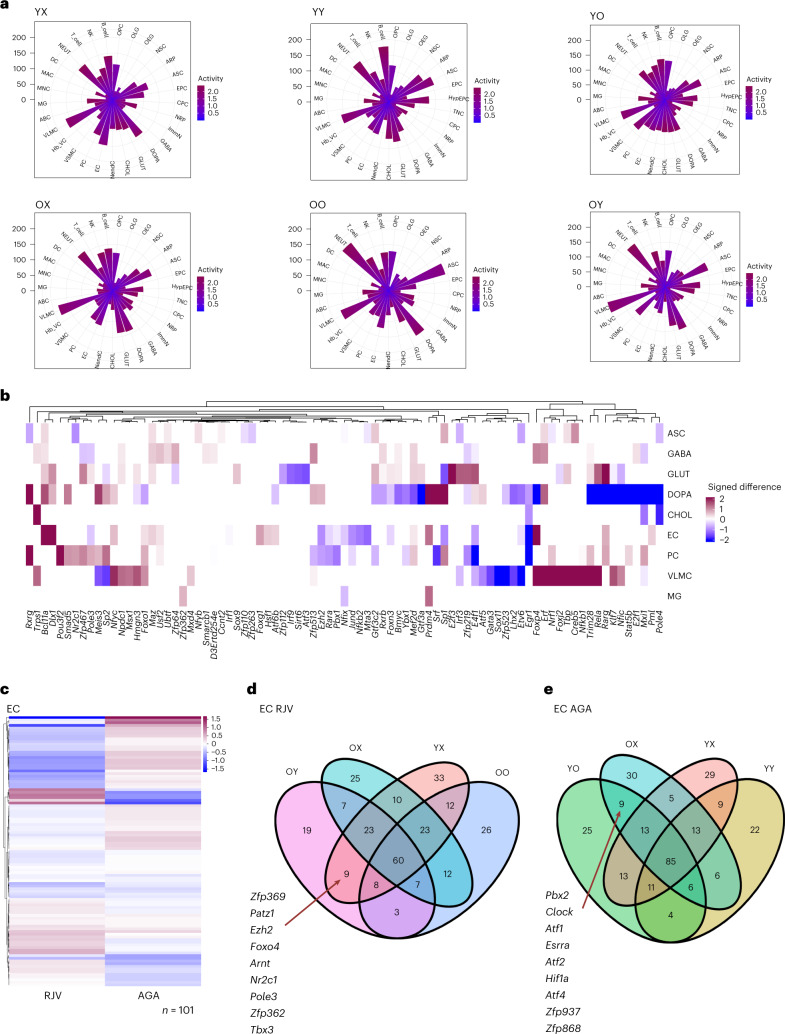
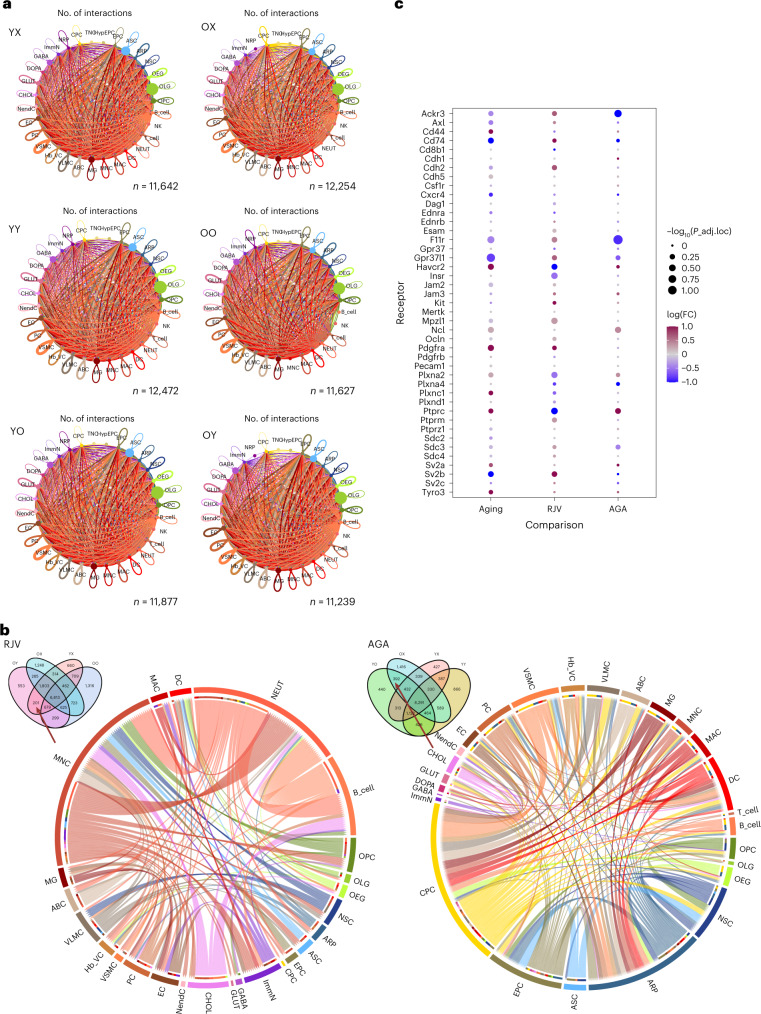
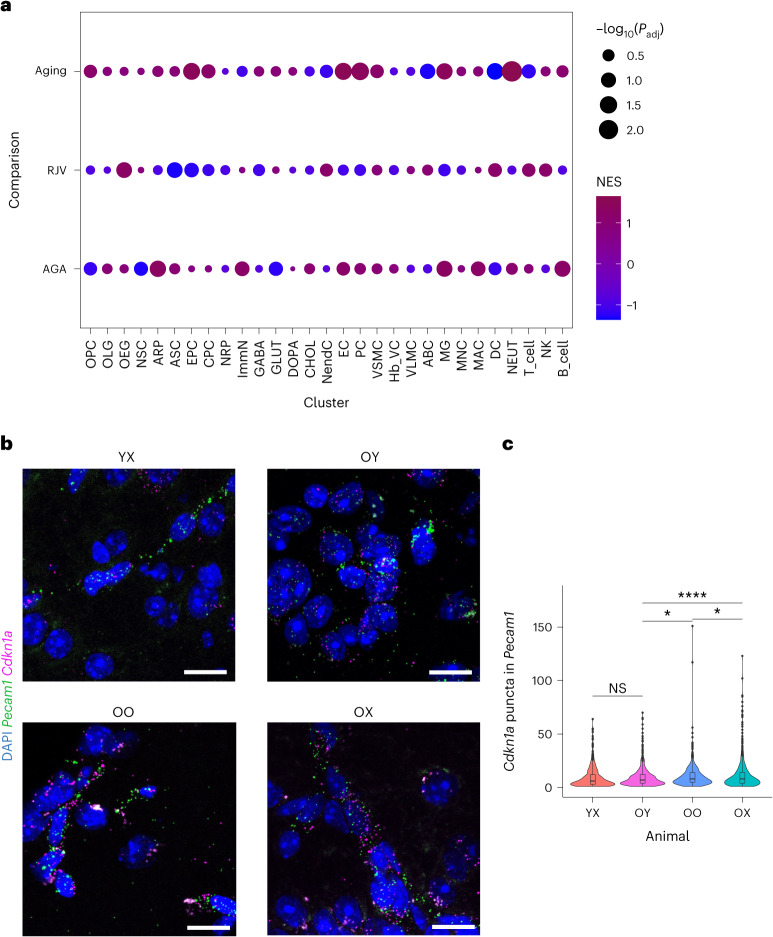
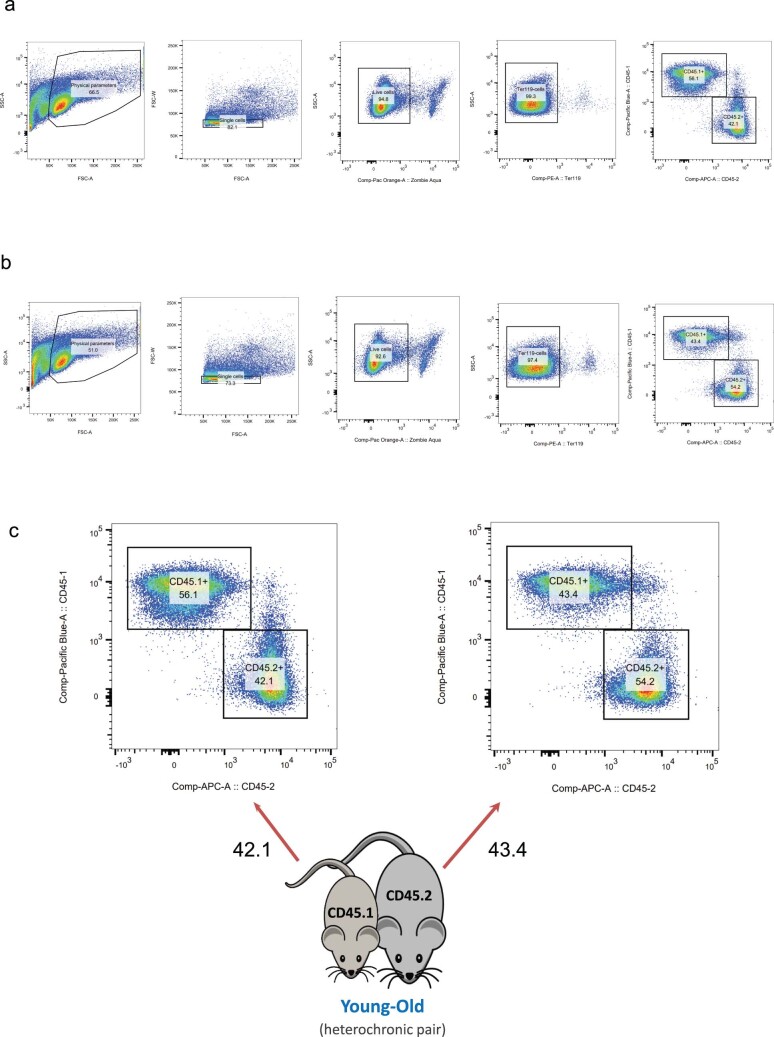
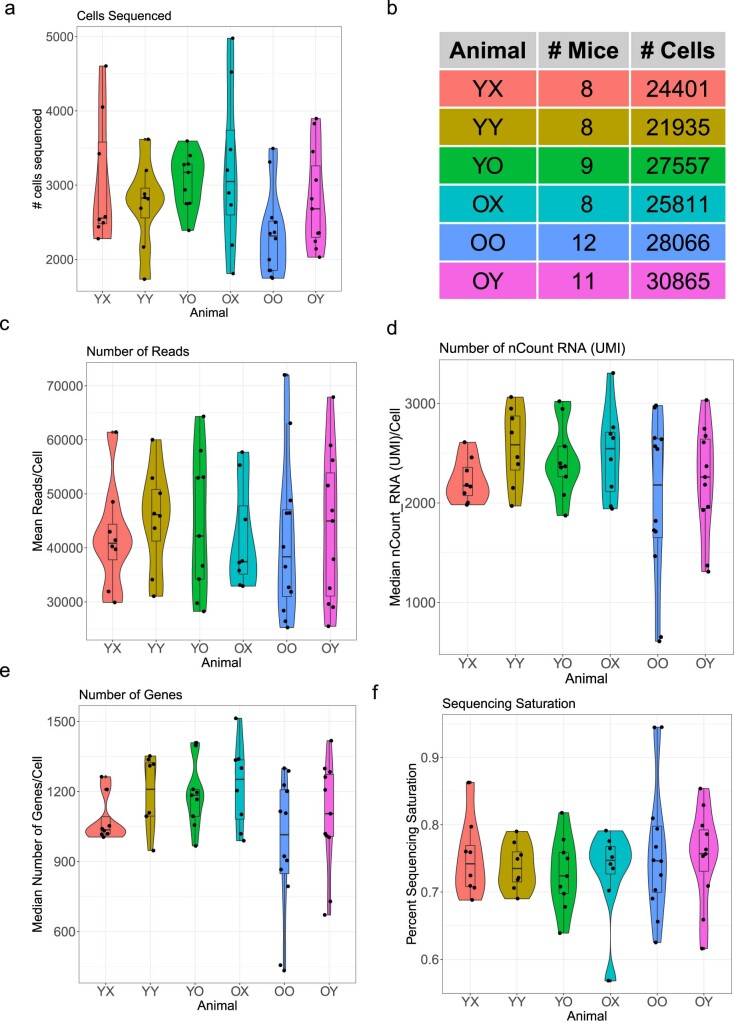
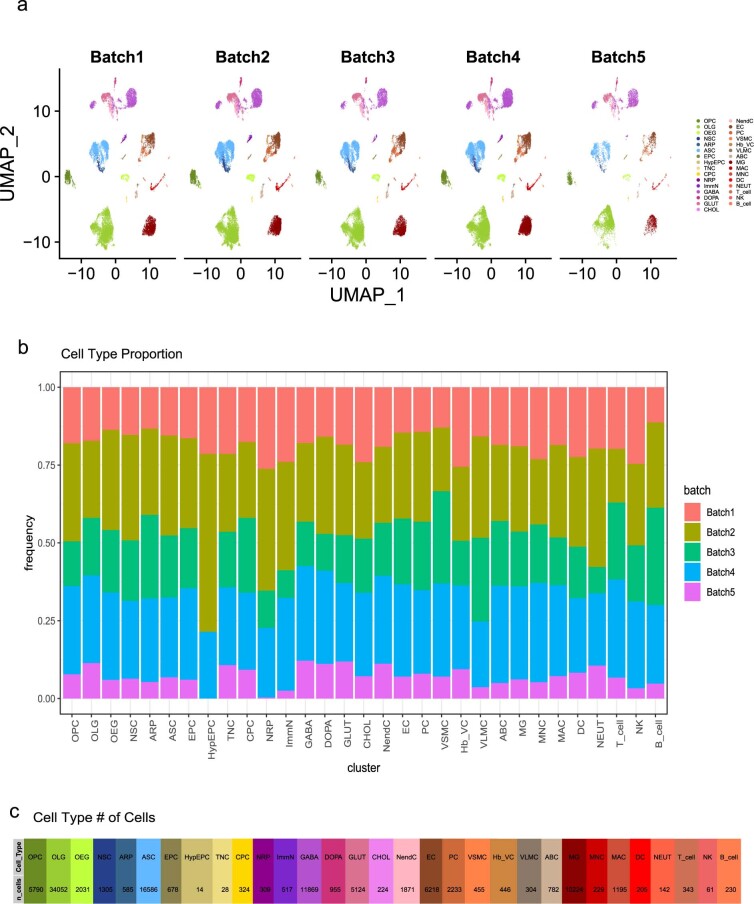
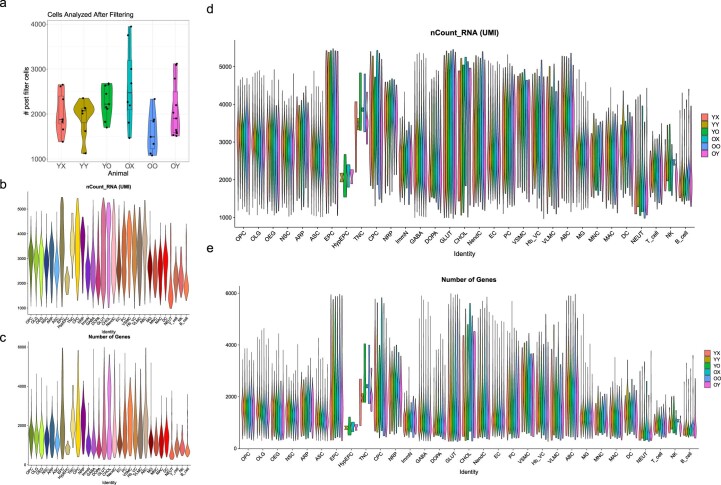
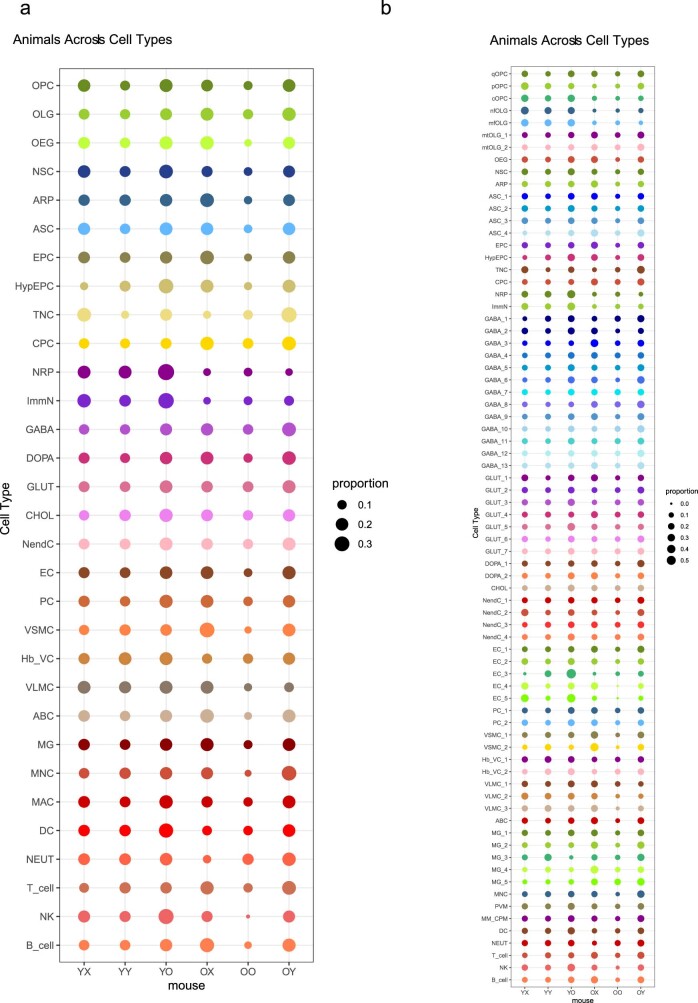
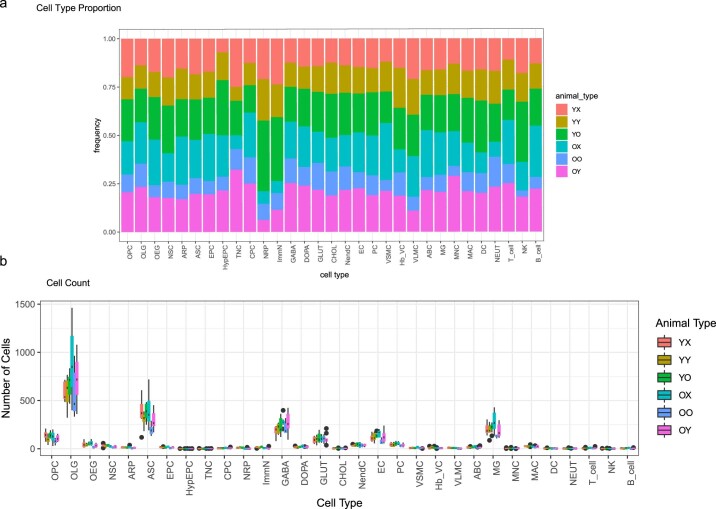
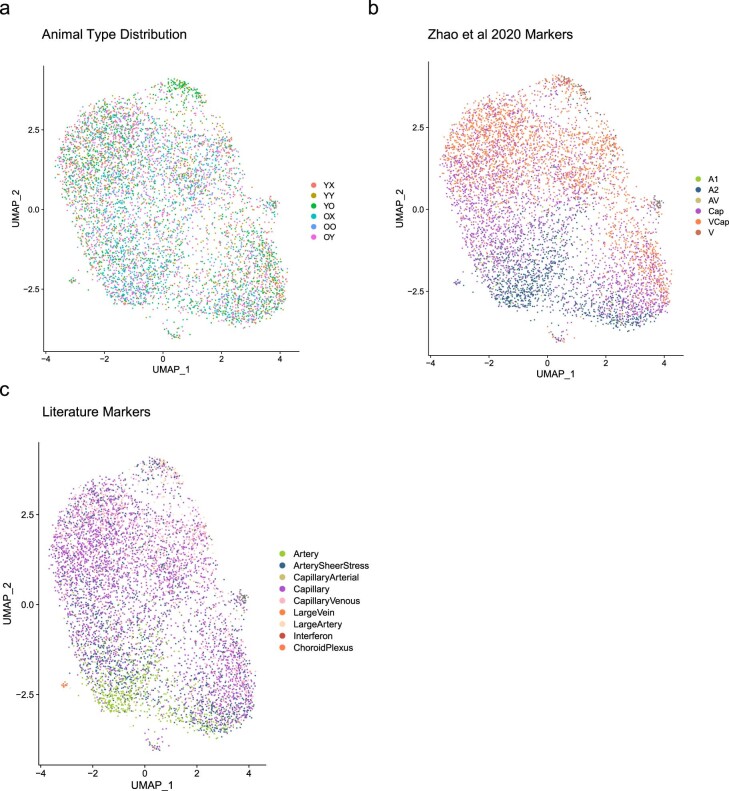
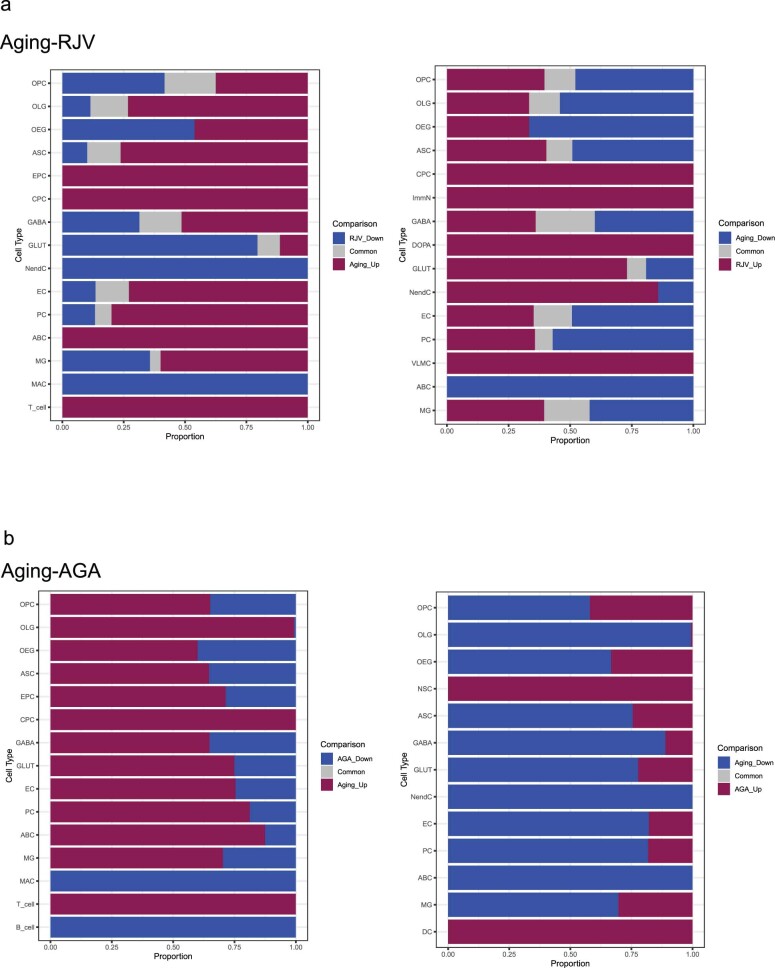
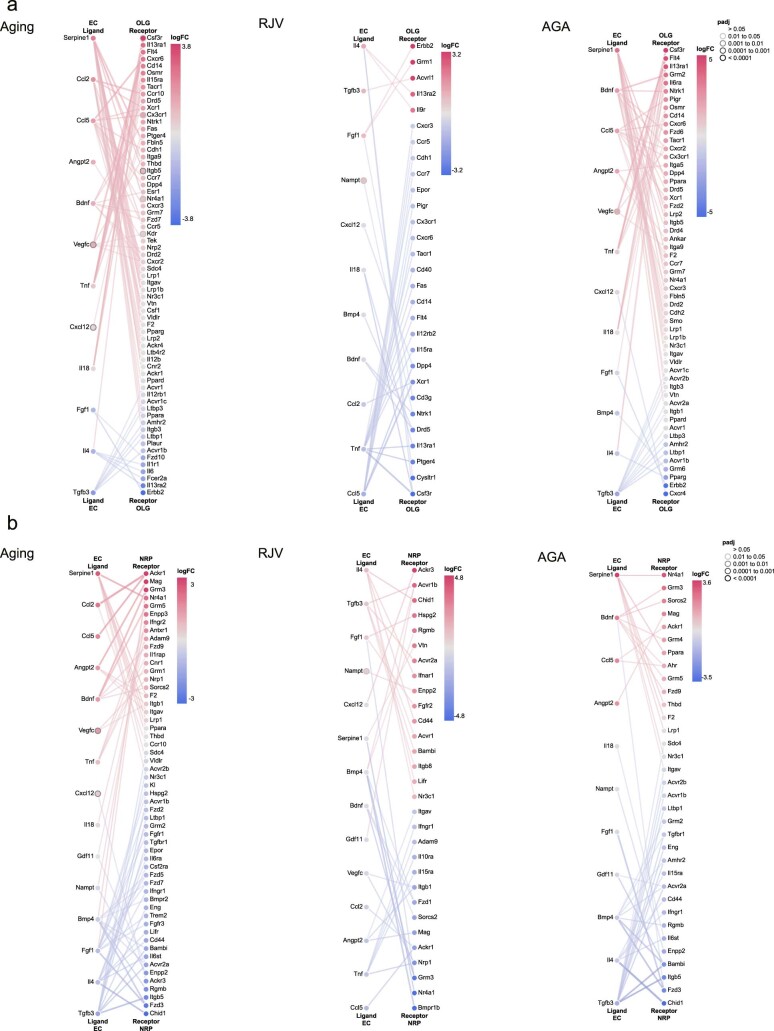
Aging is a complex process involving transcriptomic changes associated with deterioration across multiple tissues and organs, including the brain. Recent studies using heterochronic parabiosis have shown that various aspects of aging-associated decline are modifiable or even reversible. To better understand how this occurs, we performed single-cell transcriptomic profiling of young and old mouse brains after parabiosis. For each cell type, we cataloged alterations in gene expression, molecular pathways, transcriptional networks, ligand-receptor interactions and senescence status. Our analyses identified gene signatures, demonstrating that heterochronic parabiosis regulates several hallmarks of aging in a cell-type-specific manner. Brain endothelial cells were found to be especially malleable to this intervention, exhibiting dynamic transcriptional changes that affect vascular structure and function. These findings suggest new strategies for slowing deterioration and driving regeneration in the aging brain through approaches that do not rely on disease-specific mechanisms or actions of individual circulating factors.
© 2023. The Author(s).
Conflict of interest statement
L.L.R. is a founder of Elevian, Rejuveron and Vesalius Therapeutics, a member of their scientific advisory boards and a private equity shareholder. All are interested in formulating approaches intended to treat diseases of the nervous system and other tissues. He is also on the advisory board of Alkahest, a Grifols company, focused on the plasma proteome. None of these companies provided any financial support for the work in this paper. A.J.W. is a scientific advisor for Kate Therapeutics and Frequency Therapeutics, and is a founder of Elevian, Inc. and a member of their scientific advisory board and shareholder. Elevian, Inc. also provides sponsored research to the Wagers lab. A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until 31 August 2020 was a SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and Thermo Fisher Scientific. From 1 August 2020, A.R. has been an employee of Genentech, a member of the Roche Group. M.X. has been an employee of Merck & Co. since August 2020. The remaining authors declare no competing interests.
Figures
References
-
- Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell10.1016/j.cell.2022.11.001 (2022). - PubMed
-
- Burke, S. N. & Barnes, C. A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci.7, 30–40 (2006). - PubMed
-
- Navarro, A., Gomez, C., Lopez-Cepero, J. M. & Boveris, A. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am. J. Physiol. Regul. Integr. Comp. Physiol.286, R505–R511 (2004). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
