

Cell-type-specific aging clocks to quantify aging and rejuvenation in neurogenic regions of the brain
- PMID: 37118510
- PMCID: PMC10154228
- DOI: 10.1038/s43587-022-00335-4
Cell-type-specific aging clocks to quantify aging and rejuvenation in neurogenic regions of the brain
Abstract
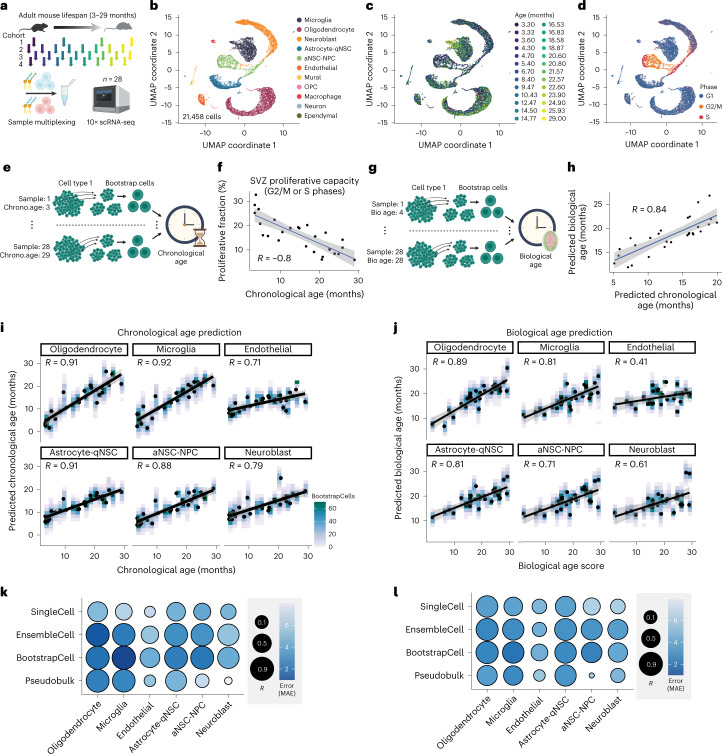
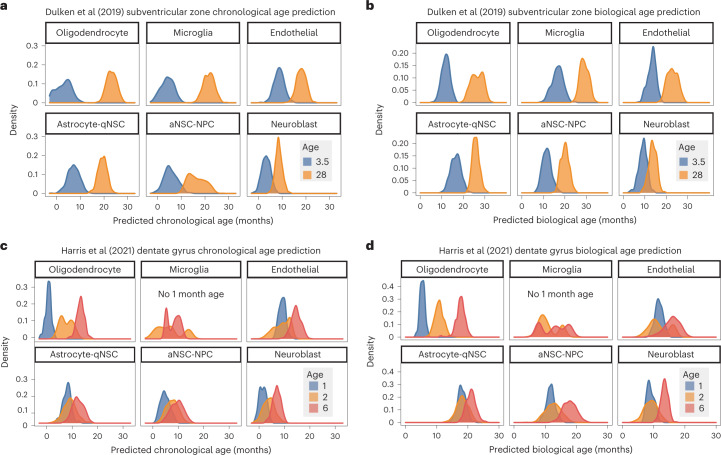
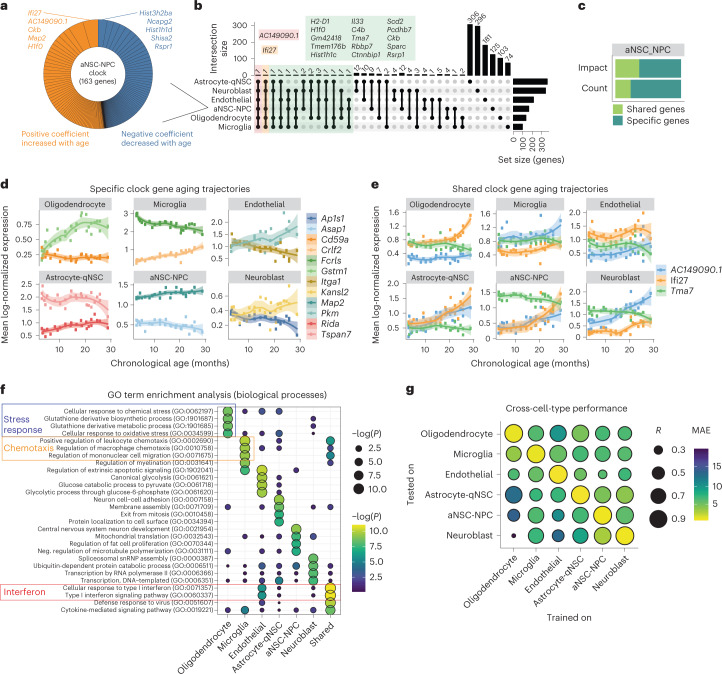
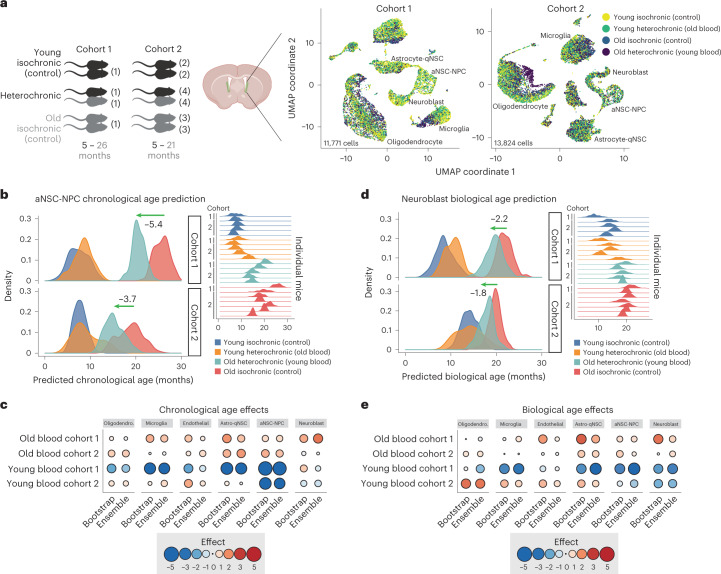
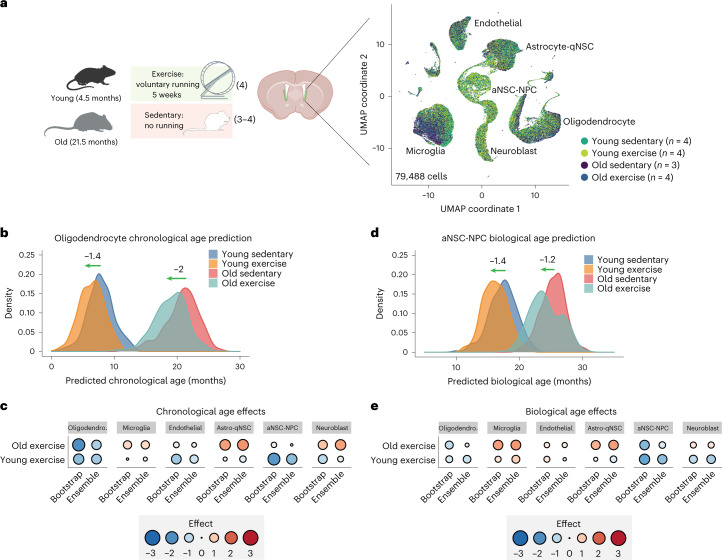
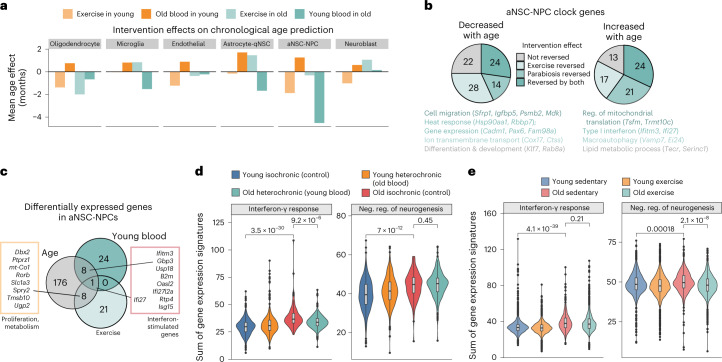
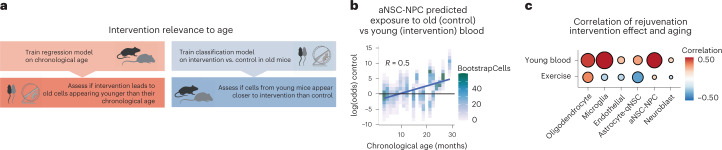
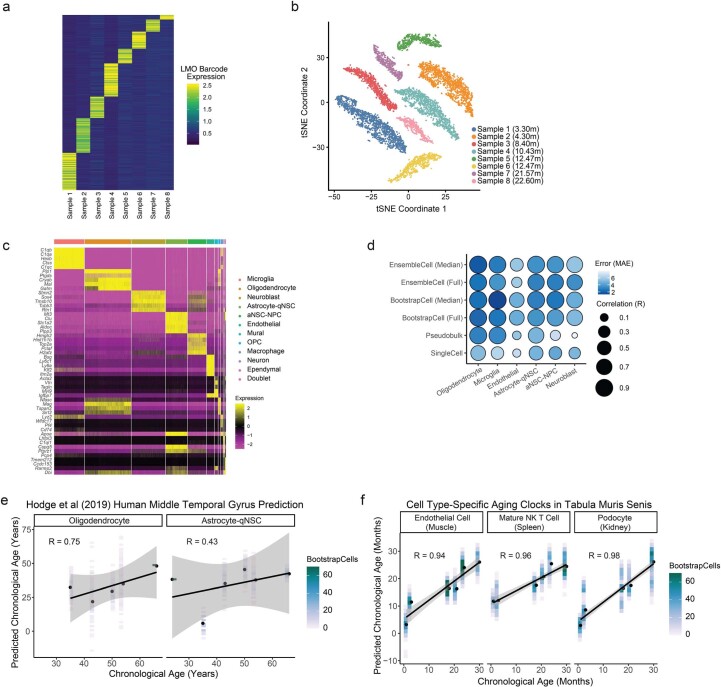
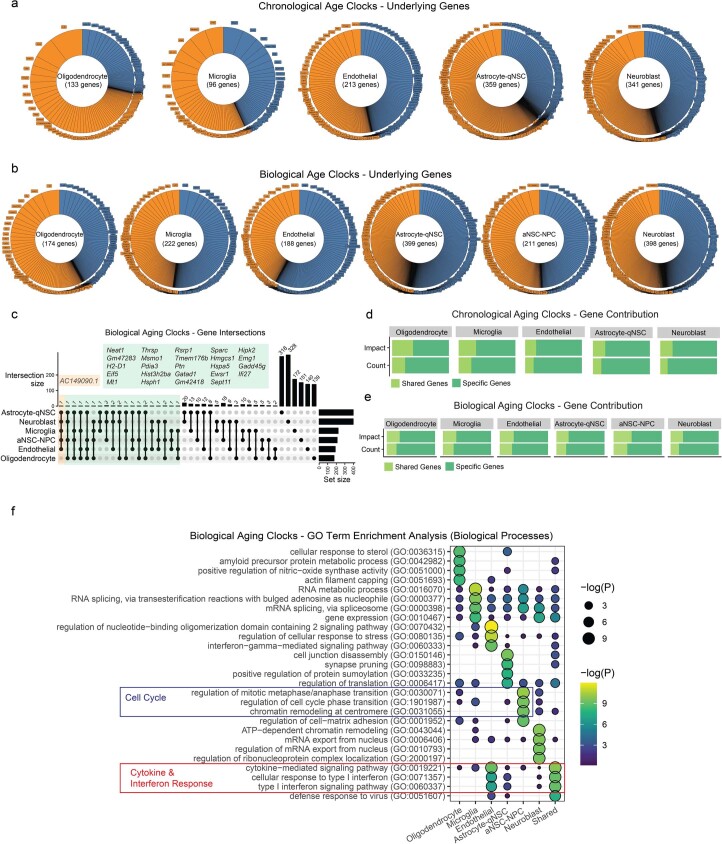
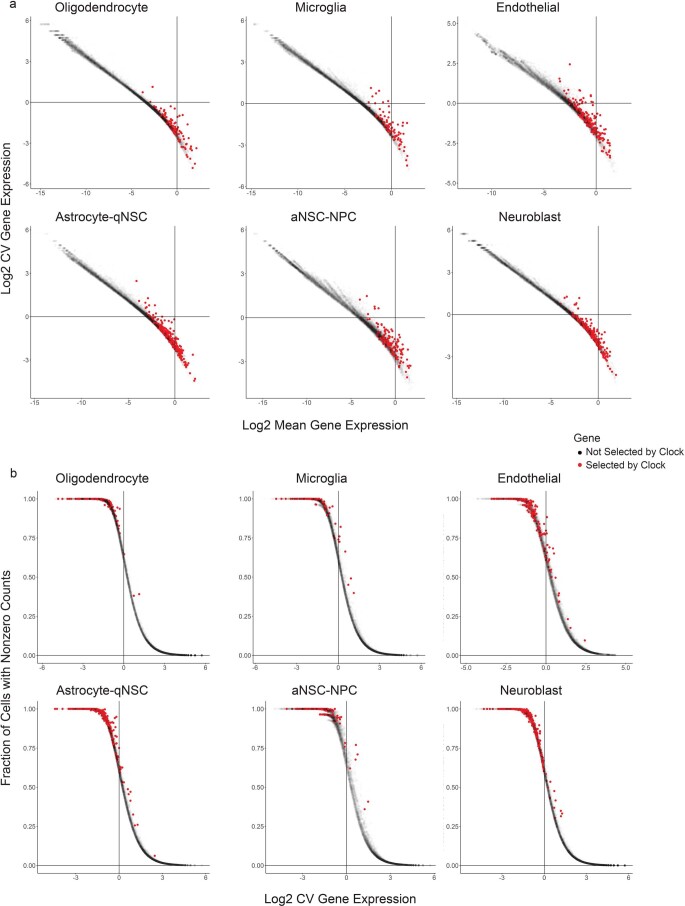
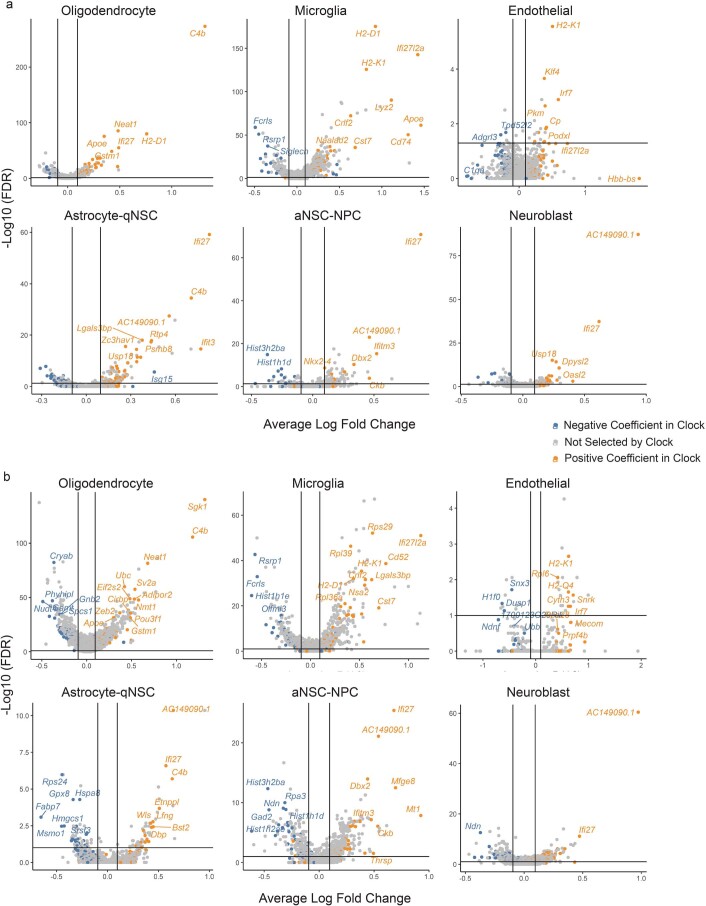
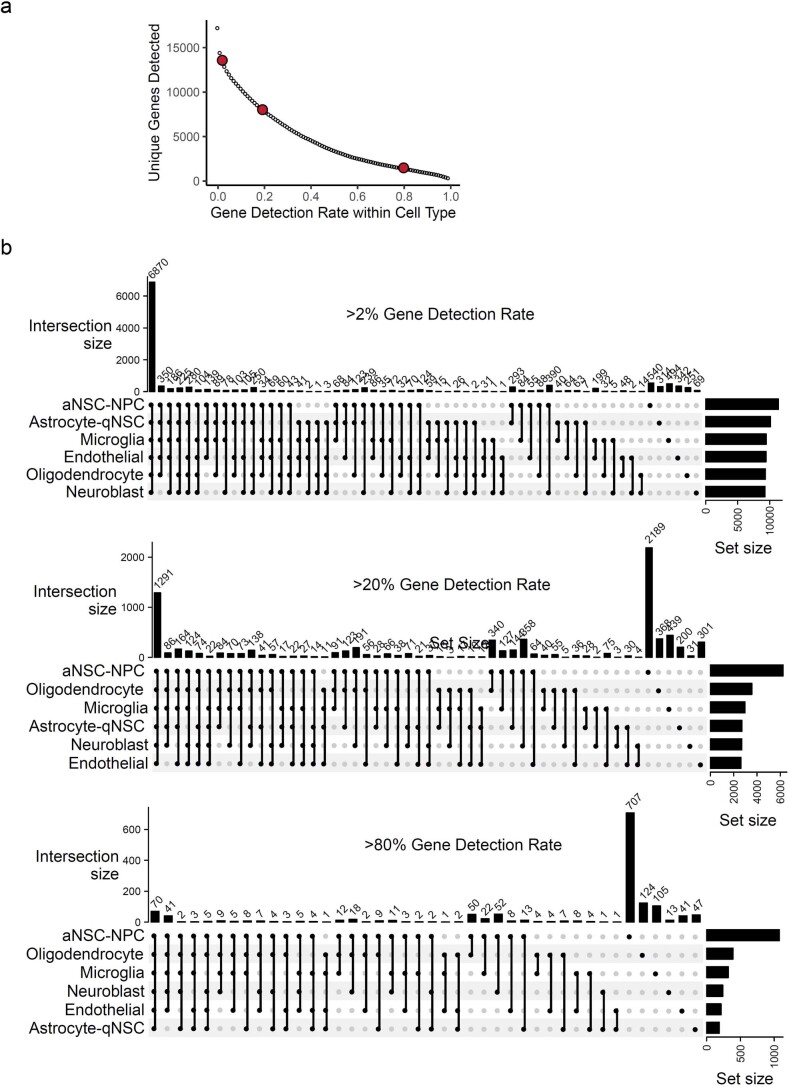
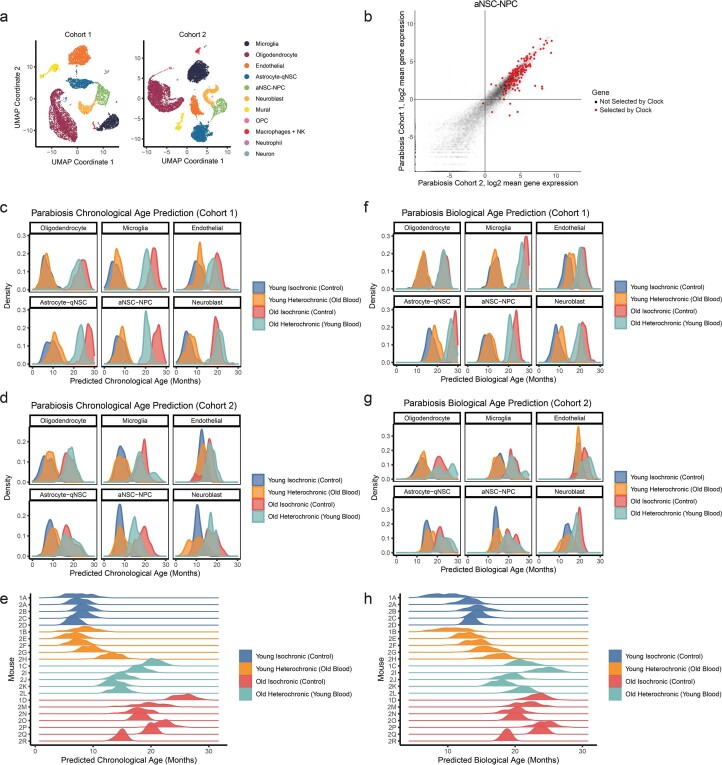
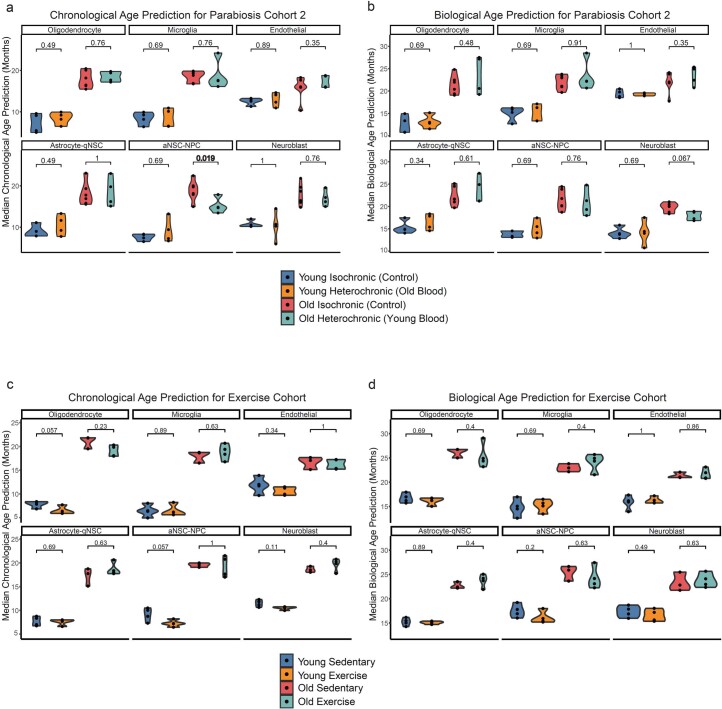
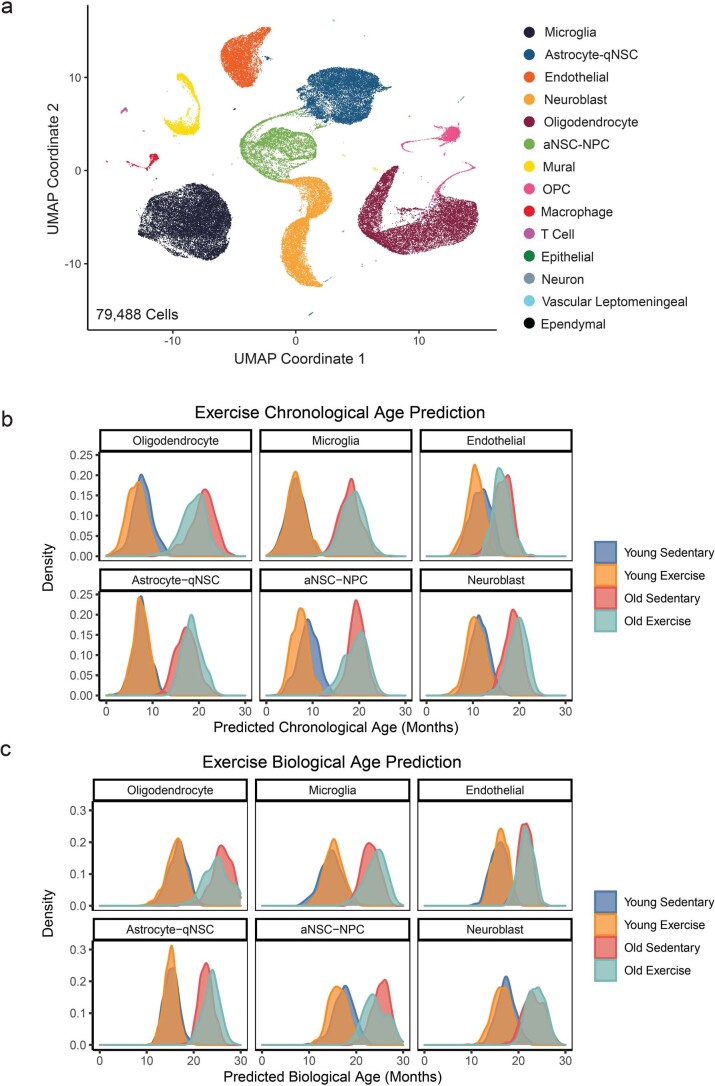
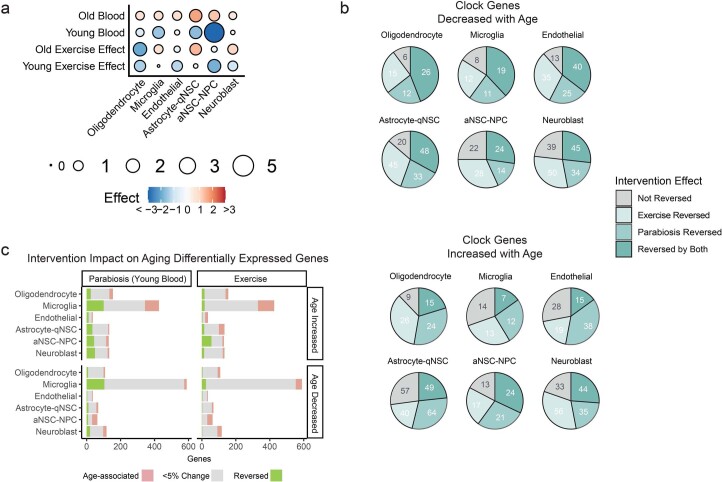
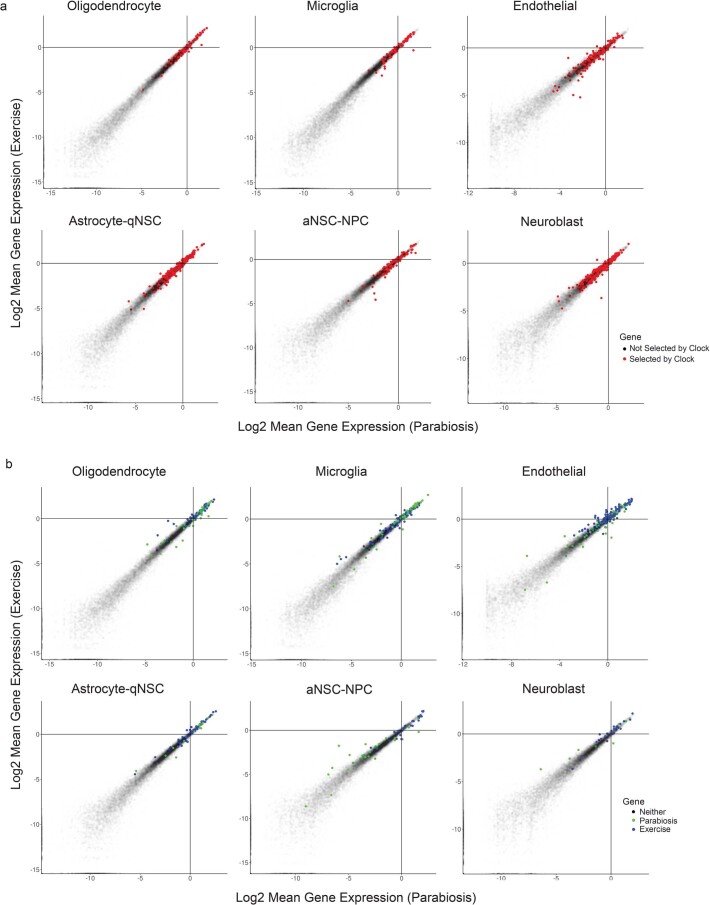
The diversity of cell types is a challenge for quantifying aging and its reversal. Here we develop 'aging clocks' based on single-cell transcriptomics to characterize cell-type-specific aging and rejuvenation. We generated single-cell transcriptomes from the subventricular zone neurogenic region of 28 mice, tiling ages from young to old. We trained single-cell-based regression models to predict chronological age and biological age (neural stem cell proliferation capacity). These aging clocks are generalizable to independent cohorts of mice, other regions of the brains, and other species. To determine if these aging clocks could quantify transcriptomic rejuvenation, we generated single-cell transcriptomic datasets of neurogenic regions for two interventions-heterochronic parabiosis and exercise. Aging clocks revealed that heterochronic parabiosis and exercise reverse transcriptomic aging in neurogenic regions, but in different ways. This study represents the first development of high-resolution aging clocks from single-cell transcriptomic data and demonstrates their application to quantify transcriptomic rejuvenation.
© 2022. The Author(s).
Conflict of interest statement
M.T.B. is a cofounder of Retro Biosciences. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
