

Inversion polymorphism in a complete human genome assembly
- PMID: 37122002
- PMCID: PMC10150506
- DOI: 10.1186/s13059-023-02919-8
Inversion polymorphism in a complete human genome assembly
Abstract
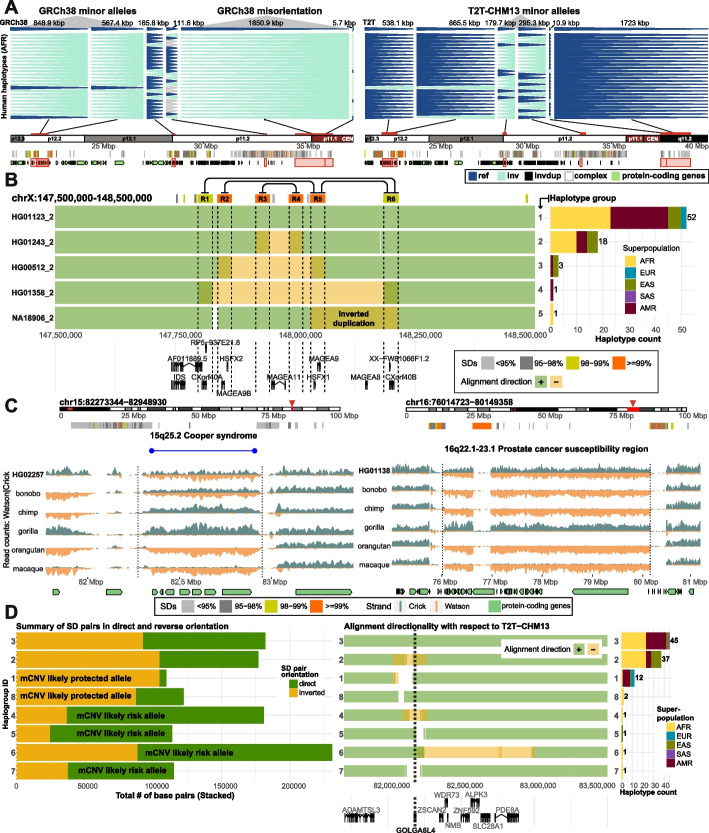
The telomere-to-telomere (T2T) complete human reference has significantly improved our ability to characterize genome structural variation. To understand its impact on inversion polymorphisms, we remapped data from 41 genomes against the T2T reference genome and compared it to the GRCh38 reference. We find a ~ 21% increase in sensitivity improving mapping of 63 inversions on the T2T reference. We identify 26 misorientations within GRCh38 and show that the T2T reference is three times more likely to represent the correct orientation of the major human allele. Analysis of 10 additional samples reveals novel rare inversions at chromosomes 15q25.2, 16p11.2, 16q22.1-23.1, and 22q11.21.
Keywords: Genomic structural variation; Inversion; Pathogenic copy number variant; Pericentromeric; T2T-CHM13.
© 2023. The Author(s).
Conflict of interest statement
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. The following authors have previously disclosed a patent application (no. EP19169090) relevant to Strand-seq: J.O.K., T.M., and D.P. The other authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
