

Clinical and genetic features of Kenny-Caffey syndrome type 2 with multiple electrolyte disturbances: A case report
- PMID: 37122511
- PMCID: PMC10131010
- DOI: 10.12998/wjcc.v11.i10.2290
Clinical and genetic features of Kenny-Caffey syndrome type 2 with multiple electrolyte disturbances: A case report
Abstract
Background: Hypoparathyroidism, which can be sporadic or a component of an inherited syndrome, is the most common cause of hypocalcemia. If hypocalcemia is accompanied by other electrolyte disturbances, such as hypokalemia and hypomagnesemia, then the cause, such as renal tubular disease, should be carefully identified.
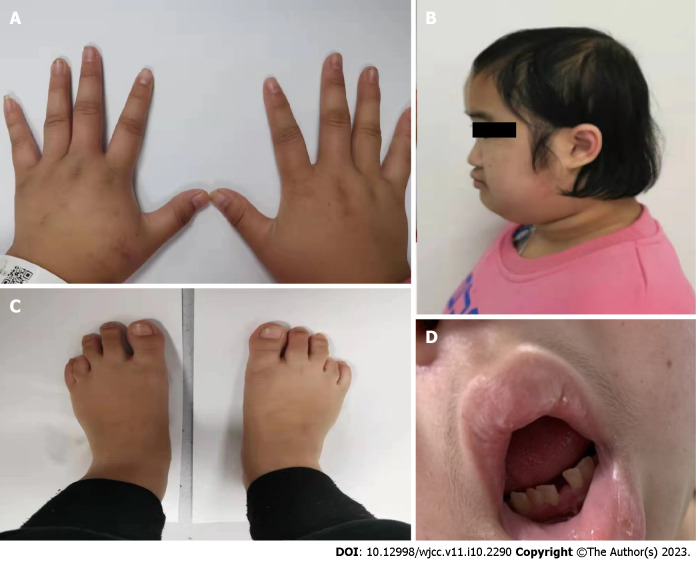
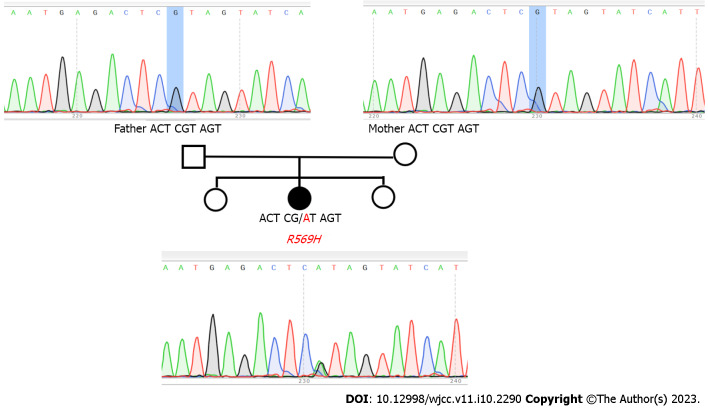
Case summary: An 18-year-old female visited our clinic because of short stature and facial deformities, including typical phenotypes, such as low ear position, depression of the nasal bridge, small hands and feet, and loss of dentition. The lab results suggested normal parathyroid hormone but hypocalcemia. In addition, multiple electrolyte disturbances were found, including hypokalemia, hypocalcemia and hypomagnesemia. The physical signs showed a short fourth metatarsal bone of both feet. The X-ray images showed cortical thickening of long bones and narrowing of the medulla of the lumen. Cranial computed tomography indicated calcification in the bilateral basal ganglia. Finally, the genetic investigation showed a de novo heterogenous mutation of "FAM111A" (c. G1706A:p.R569H). Through a review of previously reported cases, the mutation was found to be the most common mutation site in Kenny-Caffey syndrome type 2 (KCS2) cases reported thus far (16/23, 69.6%). The mutation was slightly more prevalent in females than in males (11/16, 68.8%). Except for hypocalcemia, other clinical manifestations are heterogeneous.
Conclusion: As a rare autosomal dominant genetic disease of hypoparathyroidism, the clinical manifestations of KCS2 are atypical and diverse. This girl presented with short stature, facial deformities and skeletal deformities. The laboratory results revealed hypocalcemia as the main electrolyte disturbance. Even though her family members showed normal phenotypes, gene detection was performed to find the mutation of the FAM111A gene and confirmed the diagnosis of KCS2.
Keywords: Case report; FAM111A gene; Hypocalcemia; Hypomagnesemia; Hypoparathyroidism; Kenny-Caffey syndrome type 2.
©The Author(s) 2023. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Figures

Similar articles
-
Further delineation of phenotype and genotype of Kenny-Caffey syndrome type 2 (phenotype and genotype of KCS type 2).Mol Genet Genomic Med. 2024 Apr;12(4):e2433. doi: 10.1002/mgg3.2433. Mol Genet Genomic Med. 2024. PMID: 38591167 Free PMC article. Review.
-
Kenny-Caffey Syndrome Type 2 (KCS2): A New Case Report and Patient Follow-Up Optimization.J Clin Med. 2024 Dec 28;14(1):118. doi: 10.3390/jcm14010118. J Clin Med. 2024. PMID: 39797201 Free PMC article.
-
A recurrent de novo FAM111A mutation causes Kenny-Caffey syndrome type 2.J Bone Miner Res. 2014 Apr;29(4):992-8. doi: 10.1002/jbmr.2091. J Bone Miner Res. 2014. PMID: 23996431
-
Case report: Late middle-aged features of FAM111A variant, Kenny-Caffey syndrome type 2-suggestive symptoms during a long follow-up.Front Endocrinol (Lausanne). 2023 Jan 4;13:1073173. doi: 10.3389/fendo.2022.1073173. eCollection 2022. Front Endocrinol (Lausanne). 2023. PMID: 36686468 Free PMC article. Review.
-
Expanding the Phenotypic Spectrum of Kenny-Caffey Syndrome.J Clin Endocrinol Metab. 2023 Aug 18;108(9):e754-e768. doi: 10.1210/clinem/dgad147. J Clin Endocrinol Metab. 2023. PMID: 36916904 Free PMC article.
Cited by
-
Further delineation of phenotype and genotype of Kenny-Caffey syndrome type 2 (phenotype and genotype of KCS type 2).Mol Genet Genomic Med. 2024 Apr;12(4):e2433. doi: 10.1002/mgg3.2433. Mol Genet Genomic Med. 2024. PMID: 38591167 Free PMC article. Review.
-
Kenny-Caffey Syndrome Type 2 (KCS2): A New Case Report and Patient Follow-Up Optimization.J Clin Med. 2024 Dec 28;14(1):118. doi: 10.3390/jcm14010118. J Clin Med. 2024. PMID: 39797201 Free PMC article.
References
-
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. - PubMed
-
- Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G, Harding B, Beetz R, Bilous RW, Holdaway I, Shaw NJ, Fryns JP, Van de Ven W, Thakker RV, Devriendt K. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. 2000;406:419–422. - PubMed
-
- Parvari R, Hershkovitz E, Grossman N, Gorodischer R, Loeys B, Zecic A, Mortier G, Gregory S, Sharony R, Kambouris M, Sakati N, Meyer BF, Al Aqeel AI, Al Humaidan AK, Al Zanhrani F, Al Swaid A, Al Othman J, Diaz GA, Weiner R, Khan KT, Gordon R, Gelb BD HRD/Autosomal Recessive Kenny-Caffey Syndrome Consortium. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat Genet. 2002;32:448–452. - PubMed
-
- Unger S, Górna MW, Le Béchec A, Do Vale-Pereira S, Bedeschi MF, Geiberger S, Grigelioniene G, Horemuzova E, Lalatta F, Lausch E, Magnani C, Nampoothiri S, Nishimura G, Petrella D, Rojas-Ringeling F, Utsunomiya A, Zabel B, Pradervand S, Harshman K, Campos-Xavier B, Bonafé L, Superti-Furga G, Stevenson B, Superti-Furga A. FAM111A mutations result in hypoparathyroidism and impaired skeletal development. Am J Hum Genet. 2013;92:990–995. - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
