

Scarring/arrhythmogenic cardiomyopathy
- PMID: 37125320
- PMCID: PMC10132624
- DOI: 10.1093/eurheartjsupp/suad017
Scarring/arrhythmogenic cardiomyopathy
Abstract
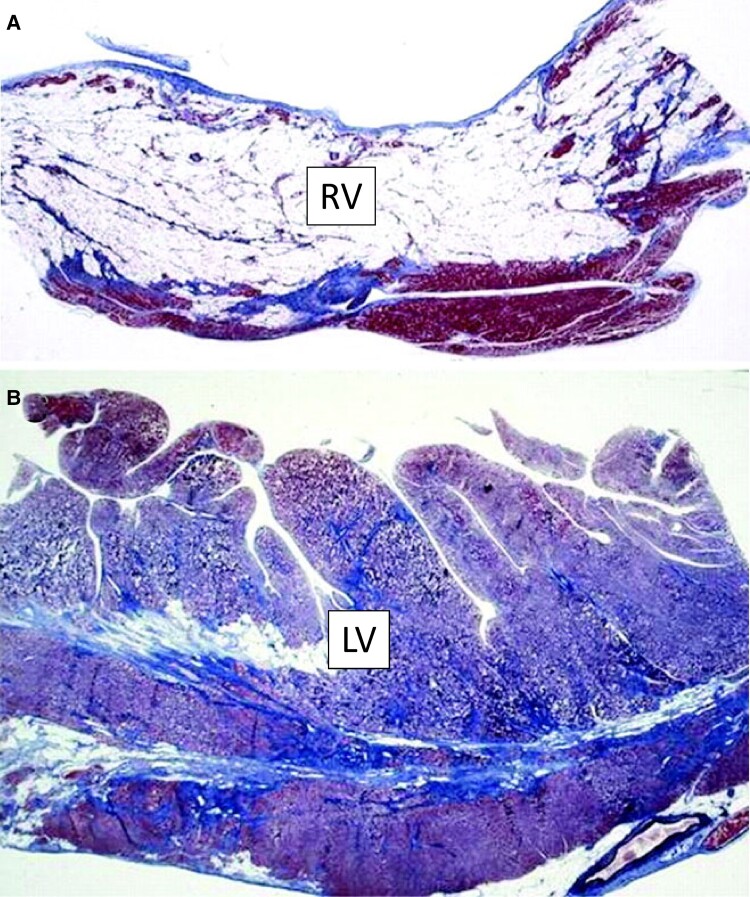
The designation of 'arrhythmogenic cardiomyopathy' reflects the evolving concept of a heart muscle disease affecting not only the right ventricle (ARVC) but also the left ventricle (LV), with phenotypic variants characterized by a biventricular (BIV) or predominant LV involvement (ALVC). Herein, we use the term 'scarring/arrhythmogenic cardiomyopathy (S/ACM)' to emphasize that the disease phenotype is distinctively characterized by loss of ventricular myocardium due to myocyte death with subsequent fibrous or fibro-fatty scar tissue replacement. The myocardial scarring predisposes to potentially lethal ventricular arrhythmias and underlies the impairment of systolic ventricular function. S/ACM is an 'umbrella term' which includes a variety of conditions, either genetic or acquired (mostly post-inflammatory), sharing the typical 'scarring' phenotypic features of the disease. Differential diagnoses include 'non-scarring' heart diseases leading to either RV dilatation from left-to-right shunt or LV dilatation/dysfunction from a dilated cardiomyopathy. The development of 2020 upgraded criteria ('Padua criteria') for diagnosis of S/ACM reflected the evolving clinical experience with the expanding spectrum of S/ACM phenotypes and the advances in cardiac magnetic resonance (CMR) imaging. The Padua criteria aimed to improve the diagnosis of S/ACM by incorporation of CMR myocardial tissue characterization findings. Risk stratification of S/ACM patients is mostly based on arrhythmic burden and ventricular dysfunction severity, although other ECG or imaging parameters may have a role. Medical therapy is crucial for treatment of ventricular arrhythmias and heart failure. Implantable cardioverter defibrillator (ICD) is the only proven life-saving treatment, despite its significant morbidity because of device-related complications and inappropriate shocks. Selection of patients who can benefit the most from ICD therapy is one of the most challenging issues in clinical practice.
Keywords: Cardiac magnetic resonance; Cardiomyopathy; Diagnosis; Sudden cardiac death; Ventricular arrhythmia.
© The Author(s) 2023. Published by Oxford University Press on behalf of the European Society of Cardiology.
Conflict of interest statement
Conflict of interest: None declared.
Figures




References
-
- Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2017;376:61–72. - PubMed
-
- Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res 2017;121:784–802. - PubMed
-
- Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996;94:983–991. - PubMed
-
- Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran Fet al. . Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 1997;30:1512–1520. - PubMed
LinkOut - more resources
Full Text Sources